Introduction
The isolation of high-quality RNA from seeds is notoriously difficult due to the high levels of polyphenols and polysaccharides found in seeds and associated mucilage of many species. The removal of phenolic compounds is important as they bind proteins and may interfere with downstream applications, as can polysaccharides and other contaminating compounds that remain. Various published extraction protocols address the issue of high levels of polyphenols, polysaccharides and secondary metabolites present in plant tissues (Chang et al., Reference Chang, Puryear and Cairney1993; Wan and Wilkins, Reference Wan and Wilkins1994; Asif et al., Reference Asif, Dhawan and Nath2000; Sharma et al., Reference Sharma, Gill and Singh2003; Birtic and Kranner, Reference Birtic and Kranner2006; Xu et al., Reference Xu, Aileni, Abbagani and Zhang2010). The hot borate method of Wan and Wilkins (Reference Wan and Wilkins1994) has been widely used for the elimination of phenolics and polysaccharides during the extraction of high-quality RNA from seeds for microarray studies (Cadman et al., Reference Cadman, Toorop, Hilhorst and Finch-Savage2006; Carrera et al., Reference Carrera, Holman, Medhurst, Dietrich, Footitt, Theodoulou and Holdsworth2008). However, despite its widespread use it is time consuming when faced with large numbers of samples. The protocol of Chang et al. (Reference Chang, Puryear and Cairney1993) developed for the extraction of RNA from pine trees employed a high salt concentration extraction buffer with cetyltrimethylammonium bromide (CTAB) and polyvinylpyrrolidone (PVP) followed by LiCl precipitation of RNA which combine to reduce polyphenol and polysaccharide carry-over. This protocol adapted by Xu et al. (Reference Xu, Aileni, Abbagani and Zhang2010) produced a protocol economical on both reagents and time. We report modifications to the protocol of Xu et al. (Reference Xu, Aileni, Abbagani and Zhang2010) that retain its speed, economy and single precipitation step while further reducing the presence of polyphenolic and polysaccharide contaminants in the resulting RNA.
Materials and methods
Seed material
Seeds of the Arabidopsis thaliana ecotype Cape Verdi Island (Cvi) (glasshouse grown) were treated in two ways. (A) Seeds were hydrated on water for 24 h at 20°C in boxes (12 × 8 cm) (Stewart Plastics Ltd, UK) containing two sheets of 3M chromatography paper; seeds were then removed from germination boxes and surface-dried with filter paper. (B) Seeds were buried in the field in October at a depth of 5 cm for 1 month and recovered as described previously (Footitt et al., Reference Footitt, Douterelo-Soler, Clay and Finch-Savage2011). In both cases, aliquots of 50–125 mg of seeds were placed in 2 ml Eppendorf tubes, frozen in liquid nitrogen and stored at –80°C until extracted. Seeds of other members of the Brassicaceae (Brassica olearacea, the double haploid Chinese kale var. alboglabra, A12DHd) were grown in a glasshouse at 16/22°C (night/day temperature) and stored at –20°C soon after harvest (Awan et al., Reference Awan, Footitt and Finch-Savage2018) (80 mg samples) and Sinapis arvensis (Herbiseed, UK) (125 mg samples) were hydrated as in (A) above.
Solutions and reagents
All plasticware and reagents were certified nuclease free. Fifty per cent PEG 20000 solution was obtained from Sigma. CTAB and NaCl or KCl were mixed together before adding liquid, as this aids solubility. β-Mercaptoethanol was added immediately prior to use and the buffer was then heated to 65°C. Extractions were performed in 2 ml Eppendorf tubes in a fume cupboard. The protocol was for up to 125 mg hydrated seeds. For larger quantities, volumes should be adjusted appropriately.
RNA extraction buffer A: 100 mM TRIS-HCl pH 8.0, 25 mM EDTA, 2 M NaCl, 2% (w/v) CTAB, 2% (v/v) PEG 20,000, nuclease free water and 2% (v/v) β-mercaptoethanol.
RNA extraction buffer B: As in (A) but 2 M KCl replaces NaCl.
Other solutions required are chloroform, 5 M NaCl and 75% ethanol. For RNA precipitation, isopropyl alcohol and a solution of 1.2 M NaCl/0.8 M Tri-Na citrate dehydrate are required.
RNA extraction protocol
The protocol is as follows:
Add 0.45 ml hot extraction buffer to the sample and homogenize immediately using a pellet pestle attached to an electric drill. Immediately add another 0.45 ml of hot extraction buffer, mix and incubate in a water bath or heating block at 65°C for 15 min only. Longer incubation times are detrimental. Place samples on ice. Add 0.5 ml of chloroform, mix well (vortex briefly) and centrifuge at 12,000 r.p.m./4°C/10 min. Transfer upper supernatant to new tube and add 0.133 ml 5 M NaCl and 0.4 ml of chloroform; mix well and centrifuge at 12,000 r.p.m./4°C/10 min. Transfer upper supernatant to new tube and repeat the previous step.
Collect upper supernatant and add to fresh tube. Add half volume of isopropanol and half volume of 1.2 M NaCl/0.8 M tri-Na citrate dehydrate; mix gently and store at room temperature for 15 min. Recover RNA by centrifugation at 12,000 r.p.m./4°C/10 min. Carefully discard the supernatant and wash the pellet with ice-cold 75% ethanol; centrifuge as above and remove ethanol. Air dry pellet for 10 min and re-dissolve in 120 µl nuclease free water and store at –80°C.
RNA analysis
The quantity and quality of RNA was determined by measuring the absorbance at 230, 260 and 280 nm using a Nanodrop spectrophotometer (Thermo Scientific, UK). Quality was evaluated by comparing A260/A280 ratio (protein) and A260/A230 ratio (polysaccharide). The quality of RNA extracted with buffer B was also determined using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, USA). Arabidopsis data were subjected to analysis of variance to determine significant differences in 260/230 ratios and RNA yields.
Arabidopsis RNA was treated with RNase-free DNase I (Roche Diagnostics) to remove contaminating genomic DNA prior to cDNA synthesis and QPCR which was performed as described elsewhere (Footitt et al., Reference Footitt, Douterelo-Soler, Clay and Finch-Savage2011, Reference Footitt, Müller, Kermode and Finch-Savage2015). Interference by genomic DNA during QPCR was further guarded against by using intron spanning primer pairs designed using the Roche Life Science Assay Design Center website (https://lifescience.roche.com/en_gb/brands/universal-probe-library.html#assay-design-center). Reference genes used for normalization of the data were At4g34270 (Tip41-like) and At4g12590 (see Footitt et al., Reference Footitt, Müller, Kermode and Finch-Savage2015). Expression of the following genes whose proteins are involved in dormancy regulation was determined in Arabidopsis seeds: CYP707A2 (At2g29090), DOG1 (At5g45830), GA2ox2 (At1g30040), GA3ox1 (At1g15550), MFT (At1g18100), NCED6 (At3g24220). For primer sequences, see Footitt et al. (Reference Footitt, Douterelo-Soler, Clay and Finch-Savage2011). Data are presented as the ratio of the geometric mean of the gene of interest/geometric mean of both reference genes.
Results and Discussion
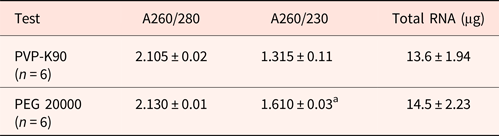
Previously, we found that commercially available RNA extraction kits produced low RNA yields and low A260/230 ratios, indicating contamination with phenolics and polysaccharides. This is problematic when the quantity and availability of seeds is limited. Traditionally, poplyvinylpyrrolidone (PVP) reduces the polyphenol contamination during RNA extraction, whereas high molecular weight polyethylene glycol (PEG) reduces both polyphenolic and polysaccharide contamination of RNA (Gehrig et al., Reference Gehrig, Winter, Cushman, Borland and Taybi2000). We therefore compared the effect of PVP and PEG 20000 on the resulting quality of RNA from hydrated Arabidopsis seeds extracted using extraction buffer A incorporating 2% PVP-K90 or 2% PEG 20000. The resulting RNA yields were similar, but the A260/230 ratio was significantly higher, indicating that PEG 20000 was more efficient in reducing polyphenol and polysaccharide contamination (Table 1).
Table 1. Comparing the impact of polyvinylpyrrolodone and polyethylene glycol on polyphenol and polysaccharide contamination of RNA
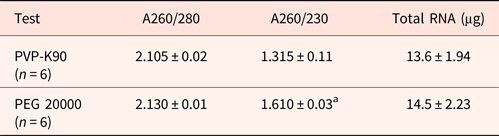
PVP-K90, polyvinylpyrrolodone; PEG 20000, polyethylene glycol. RNA was extracted from unburied Arabidopsis seeds using buffer A. Polyphenol and polysaccharide contamination was as reflected by the 260/230 ratio. Arabidopsis, mean values for A260/230 and total RNA followed by letter “a” are significantly different (P < 0.05).
The RNA pellets using this protocol (buffer A) were still gelatinous, indicating the presence of polysaccharides. Additional post-extraction steps to precipitate polysaccharides include precipitation with 3 M sodium acetate (pH 5.2) and ethanol (Asif et al., Reference Asif, Dhawan and Nath2000). However, the introduction of additional steps potentially reduces yield and increases the extraction time. To address this, we looked at alternative protocols for the precipitation of polysaccharides. High NaCl concentrations have been proposed as a method for polysaccharide removal from DNA (Fang et al., Reference Fang, Hammar and Grumet1992). However, KCl is a more efficient precipitant of both neutral and acidic polysaccharides. The efficiency of this increases under alkaline conditions (ionization of acidic polysaccharides) and in the presence of ethanol as alcohols reduce polysaccharide solubility (Smidsrød and Haug, Reference Smidsrød and Haug1967; Smidsrød et al., Reference Smidsrød, Larsen, Pernas and Haug1967). CTAB is an effective precipitant of acidic polysaccharides (Bera et al., Reference Bera, Foster and Stacey1955; Smidsrød and Haug, Reference Smidsrød and Haug1967; Murray and Thompson, Reference Murray and Thompson1980). We therefore modified the extraction buffer by replacing NaCl with KCl to increase the efficiency of polysaccharide precipitation to produce RNA extraction buffer B. The presence of β-mercaptoethanol may well further increase the efficiency of KCl.
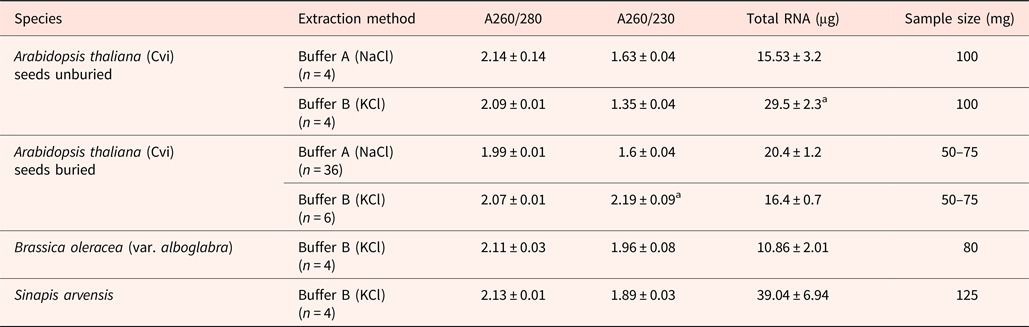
When buried Arabidodpsis seeds were extracted with buffer A (containing NaCl) the A260/230 ratio was 1.6; this increased to 1.88 ± 0.04 following precipitation with a half volume of 5 M NH4oAc and 2.5 volumes of ethanol at –20°C for at least 1 h, but this did not remove the gelatinous matrix. Only extraction with buffer B (containing KCl) achieved this, resulting in a fragile white RNA pellet and eliminating the NH4oAc precipitation step. Using buffer B significantly increased RNA yields, but not the A260/230 ratio when unburied seeds still bearing mucilage were extracted. In the case of buried Arabidopsis seeds where the mucilage coat is lost, RNA yield was not increased, but the A260/230 ratio was greater than 2. Similarly, when Brassica oleracea and Sinapis arvensis seeds were extracted the A260/230 ratio approached 2.0, resulting in a fragile white RNA pellet (Table 2). The addition of KCl to the extraction buffer effectively removes polysaccharides and other contaminants prior to the final RNA precipitation step, whereas they would normally co-precipitate in the RNA precipitation buffer.
Table 2. Comparison of RNA extraction with NaCl- and KCl-containing buffers from Arabidopsis and other species
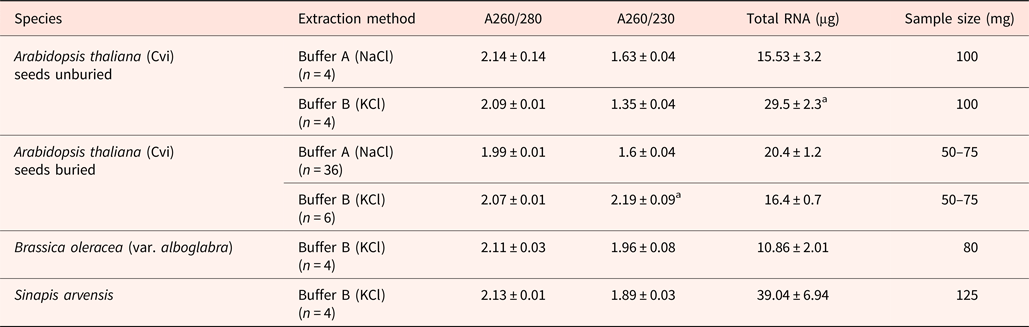
Extraction used buffer A (NaCl-containing) and buffer B (KCl-containing). Data from buried Arabidopsis seeds extracted with buffer A are from RNA prior to precipitation with a half volume of 5 M NH4oAc and 2.5 volumes of ethanol at –20°C for at least 1 h for RT-QPCR in Footitt et al. (Reference Footitt, Huang, Clay, Mead and Finch-Savage2013). In Arabidopsis, mean values for A260/230 and total RNA followed by letter “a” are significantly different (P < 0.05).
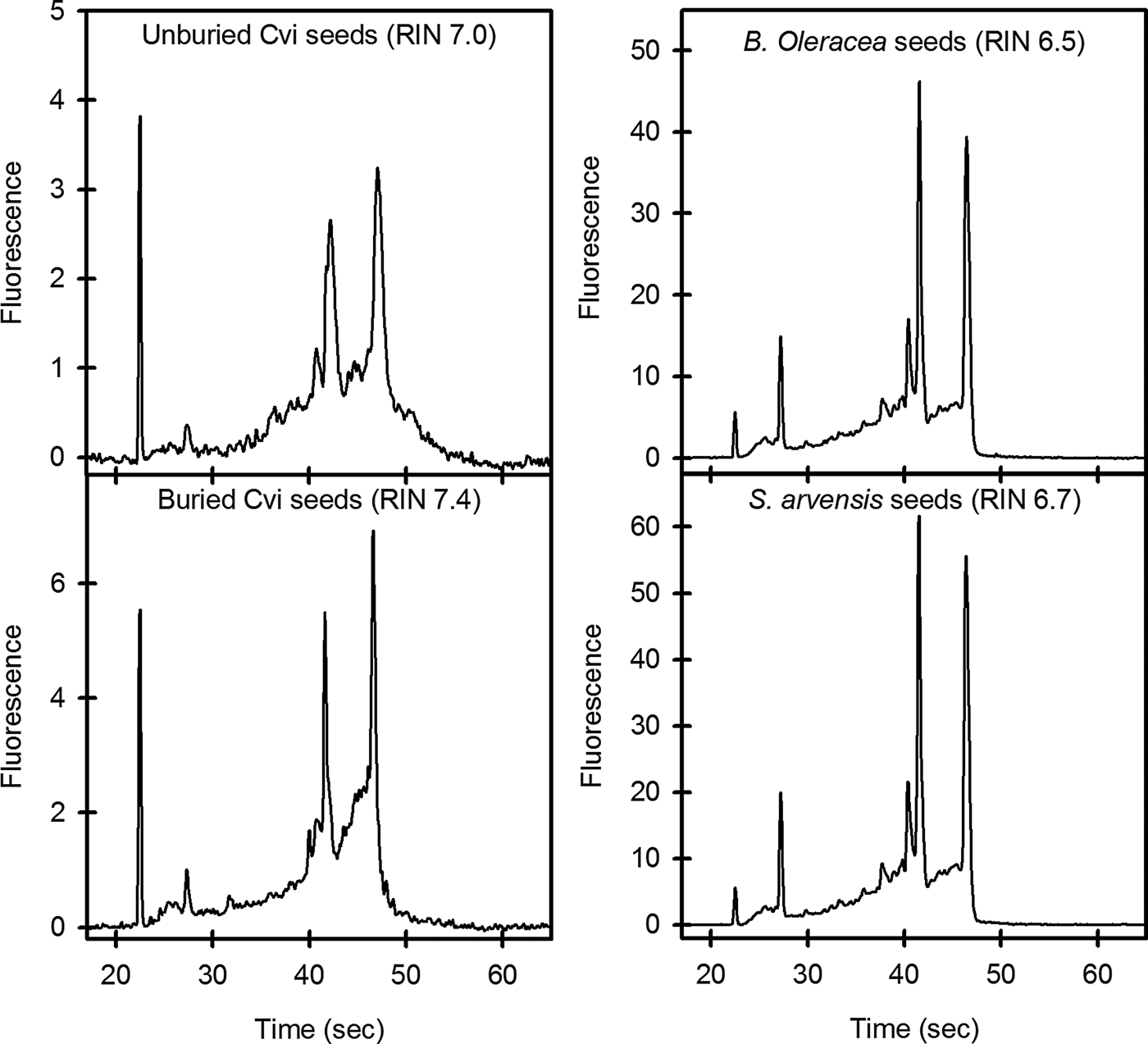
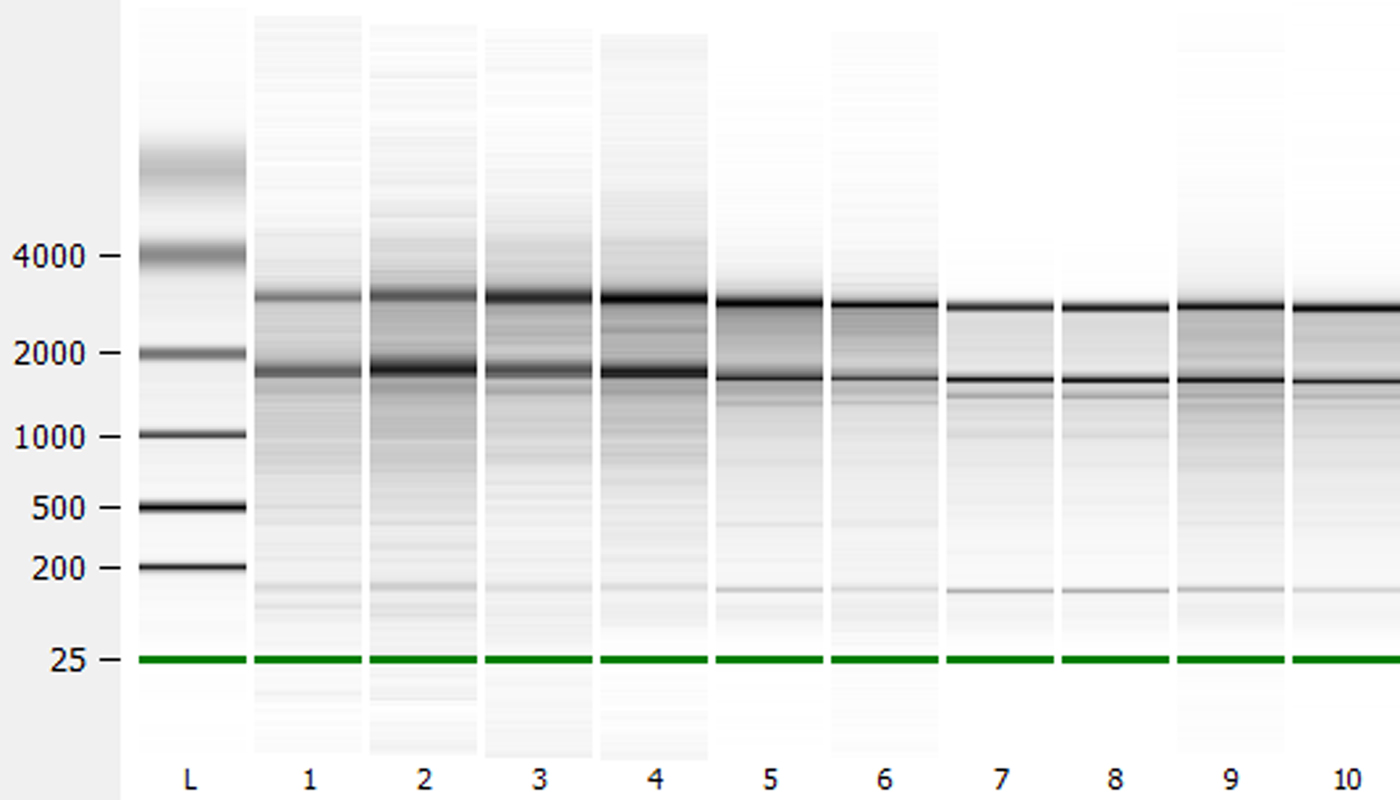
Further analysis of RNA quality using an Agilent 2100 Bioanalyzer produced RNA integrity numbers (RIN values; 1 = poor quality, 10 = high quality) of 6.4–7.0 for unburied Arabidopsis and 7.2–7.5 for Arabidopsis recovered from the field soils. Brassica oleracea and Sinapis arvensis had values ranging from 6.4 to 6.7 and 6.4 to 6.5, respectively. When electropherograms were compared for RNA from unburied and buried Arabidopsis seeds, increased background signal was seen in the unburied sample, reflecting the level of polysaccharide remaining in these samples (Fig. 1). Electropherograms of B. oleracea and S. arvensis RNA are similar to that of buried Arabidopsis seeds, reflecting their low level of polysaccharide. Gel images generated from these electropherograms also reflect this reduced contamination level (Fig. 2). The yield of RNA was within the range reported previously for Arabidopsis seeds (Vicient and Delseny, Reference Vicient and Delseny1999; Birtic and Kranner, Reference Birtic and Kranner2006). Results similar to those of Birtic and Kranner (Reference Birtic and Kranner2006) were obtained for the reduction in polysaccharide contamination for buried Arabidopsis seeds, and for B. oleracea and S. arvensis. However, the advantage of the protocol outlined here is the speed with which large numbers of samples can be processed. The Arabidopsis RNA produced was of suitable quality for downstream application such RT-QPCR (Fig. 3). The pattern of gene expression shown in the example is representative of seeds in low dormancy prior to burial, i.e. relatively high CYP707A2 expression (ABA catabolism) and low NCED6 expression (ABA synthesis). The buried seeds show the reverse pattern of expression consistent with induced deep dormancy following burial (Footitt et al., Reference Footitt, Douterelo-Soler, Clay and Finch-Savage2011).
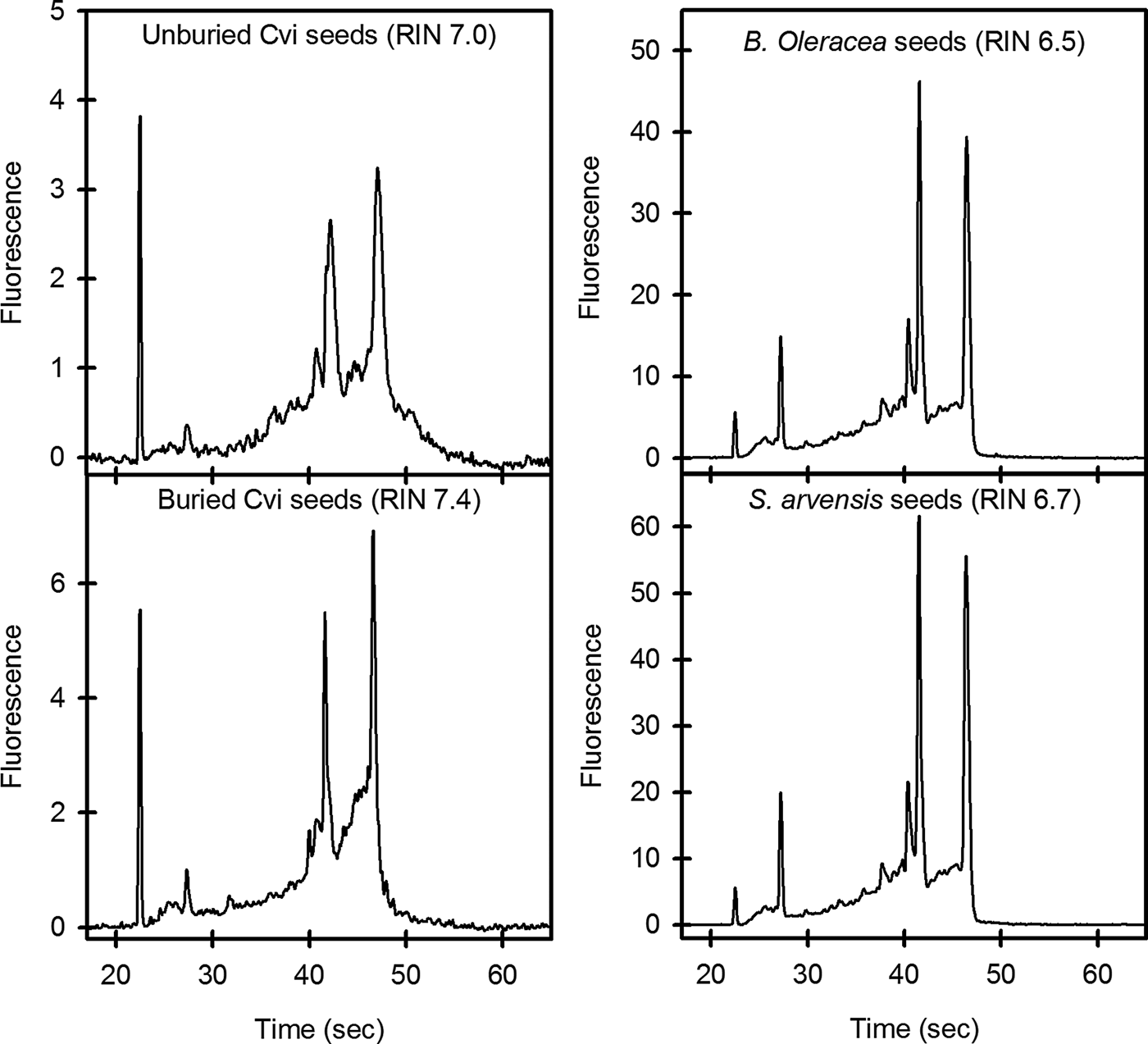
Figure 1. Electropherograms of RNA extracted from seeds with buffer B (KCl-containing). Electropherograms of Arabidopsis Cvi seeds used 40 ng μl–1 of RNA, and those of B. oleracea and S. arvensis used 200 ng μl–1 of RNA.
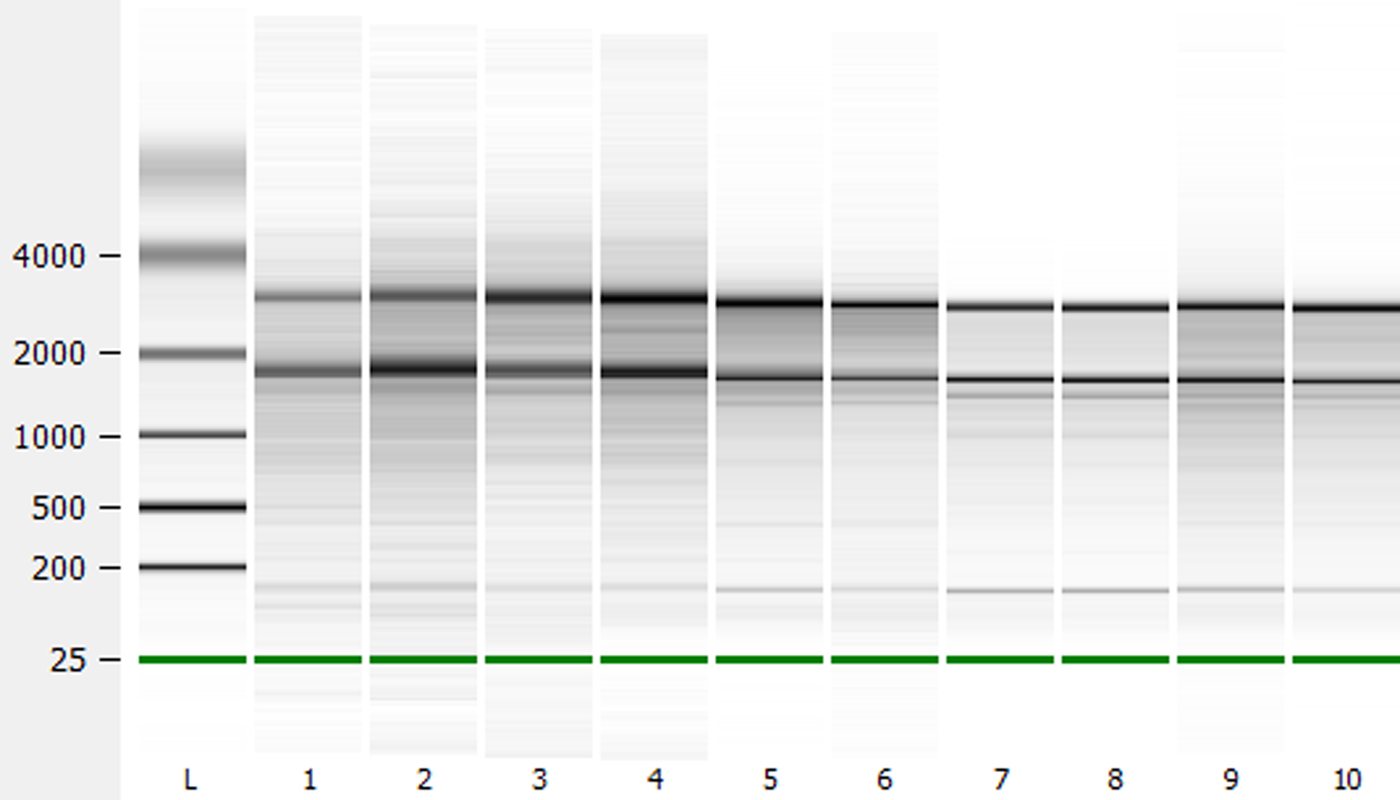
Figure 2. Electropherogram generated gel image of RNA from Arabidopsis, B. oleracea and S. arvensis seeds extracted with buffer B (KCl-containing): Ladder (L), unburied Arabidopsis Cvi seeds (1–3), buried Arabidopsis Cvi seeds (4–6), B. oleracea seeds (7–8) and S. arvensis seeds (9–10).

Figure 3. Relative gene expression in Arabidopsis Cvi seed RNA extracted using KCl-based extraction buffer prior to (unburied) and after recovery from 1 month burial in field soils (buried). Samples are representative of seeds in low dormancy prior to burial and deep dormancy following burial as described in Footitt et al. (Reference Footitt, Douterelo-Soler, Clay and Finch-Savage2011). The genes DOG1 and MFT are involved in dormancy induction and maintenance. CYP707A2 and NCED6 genes are involved in abscisic acid catabolism and synthesis, respectively. GA2ox2 and GA3ox1 are genes of gibberellin catabolism and synthesis, respectively (see Footitt et al., Reference Footitt, Douterelo-Soler, Clay and Finch-Savage2011, Reference Footitt, Huang, Clay, Mead and Finch-Savage2013). Data are means ± standard error of the mean (n = 4).
In conclusion, we show that adaptation of the CTAB protocol to minimize polyphenols and polysaccharide contaminants in the early extraction steps retains the speed and high throughput aspects of the protocol, but also has the advantage of highly improved quality of the resultant RNA. Furthermore, we show the successful application of the protocol to seeds of other Brassica species.
Financial support
This work was supported by a grant from the Biotechnology and Biological Sciences Research Council-UK (BB/1022202/1) awarded to W.E.F.-S. and S.F. S.A. was supported by European Union Framework Programme 7-Knowledge-Based Bio Economy grant 311480 EcoSeed awarded to W.E.F.-S.
Author contributions
The protocol was devised by S.F. and experiments performed by S.F. and S.A. The manuscript was written by S.F., S.A. and W.E.F.-S.