INTRODUCTION
Soil organic nitrogen (N) accounts for 95–98% of the total soil N content (e.g. Stevenson Reference Stevenson1982; Schulten and Schnitzer Reference Schulten and Schnitzer1998), with amino acids and amino sugars identified as the major soil organic N compounds (e.g. Senwo and Tabatabai Reference Senwo and Tabatabai1998; Friedel and Scheller 2002). This amino fraction serves as a substrate for soil enzymes involved in N mineralization in soils (Vieublé Gonod et al. Reference Vieublé Gonod, Jones and Chenu2006), as an energy source for soil microorganisms and as the N source for plants and microbes (Mengel Reference Mengel1996). Gruber and Galloway (Reference Gruber and Galloway2008) highlighted the dramatic change in the Earth N cycle associated with anthropogenic N inputs to the biosphere. As soil N is the major pool on Earth, it deserves further investigations to better evaluate, both qualitatively and quantitatively, its evolution under external forcings such as climate and land-use changes.
The amino acid fraction is tightly linked to microorganism activity. Microorganisms either release amino acids, take up amino acids as intact molecules, or mineralize them to inorganic N (Geisseler et al. Reference Geisseler, Horwath, Joergensen and Ludwig2010) and α-keto acids after enzymatic deamination (Geisseler and Horwath Reference Geisseler and Horwath2014) to finally release CO2. Despite the status of protein-derived amino acids in soil as degradable molecules and labile products for microorganisms, a mean residence time of several years or even decades has already been shown (Gleixner et al. Reference Gleixner, Bol and Balesdent1999, Reference Gleixner, Poirier, Bol and Balesdent2002; Schmidt et al. Reference Schmidt, Torn, Abiven, Dittmar, Guggenberger, Janssens, Kleber, Kögel-Knaber, Lehmann, Manning, Nannipieri, Rasse, Weiner and Trumbore2011). A great part of the protein fraction of soil organic matter can be stabilized in the long term (Bol et al. Reference Bol, Poirier, Balesdent and Gleixner2009), for example, by minerals (Kleber et al. Reference Kleber, Sollins and Sutton2007). Another mechanism of stabilization is the compound recycling process by microbial biomass (external cycling), e.g. the reuse of all or part of ancient compounds (Gleixner et al. Reference Gleixner, Poirier, Bol and Balesdent2002). This reutilization could lead to an underestimation of the actual turnover dynamics.
Several studies have been performed to investigate the exchanges between inorganic N and organic N and to follow organic N dynamics in the short to medium term (Kramer et al. Reference Kramer, Doane, Horwath and van Kessel2002; Holub and Lajtha Reference Holub and Lajtha2004; Takebayashi et al. Reference Takebayashi, Koba, Sasaki, Fang and Yoh2010). Most of them, however, derived from 15N labeling experiments that cannot inform on the long-term dynamics. To counteract this limitation, the present study focused on carbons of amino acids, and more specifically on the α-carboxyl amino carbon (hereafter carboxyl carbon) to tackle both mid- and long-term dynamics, using both natural 13C (C3/C4 vegetation succession; Balesdent and Mariotti Reference Balesdent and Mariotti1996) and 14C (natural production and bomb-peak) labeling.
The aim of this investigation was to study the dynamic of carboxyl carbon of the amino acid fraction of soil from a C3/C4 vegetation succession of a 15-yr chronosequence. We analyzed the 13C and 14C signature of the carboxyl carbon of the amino acid fraction to provide a new understanding of soil organic matter dynamics.
MATERIAL AND METHODS
Soil
Samples came from the Closeaux experimental field in Versailles, France (Dignac et al. Reference Dignac, Bahri, Rumpel, Rasse, Bardoux, Balesdent, Girardin, Chenu and Mariotti2005). The soil is categorized by the United Nations Food and Agriculture (FAO) soil system as an Eutric Cambisol with a silt loam texture. This field follows the schema of a C3/C4 chronosequence initially covered by wheat (C3) that was sequentially planted with maize (C4).
Amino acid screening of the wheat reference plot highlighted a combination of biogenic amino acids, including aspartic acid, threonine, leucine, alanine, valine, glycine, glutamic acid, serine, isoleucine, and lysine in decreasing order of abundance. The isotopic value was measured on individual amino acids by gas chromatography/combustion/isotope ratio mass spectrometry of n-tertbutyldimethylsilyl amino acids according to the method developed by Kheirbeik et al. (Reference Kheirbeik, Hatté and Balesdent2016). The average δ13C of the total amino acid pool reached –27.2‰ in the wheat soil (L Kheirbeik, unpublished data from PhD thesis in progress).
Samples
We collected the upper layers (0–25 cm) of soils from parcels with 0, 3, 9, and 15 yr of C4 plants. Samples with 0, 3, and 9 yr were collected in 2002 and underwent maize cultivation as C4 plants. The sample with 15 yr of C4 plants was collected in 2008, having been under maize from 1993 to 2004, then sorghum in 2005, 2006, and 2007. Soil samples were air-dried, then sieved at 2 mm, discarding coarse plant residues.
Laboratory Procedure
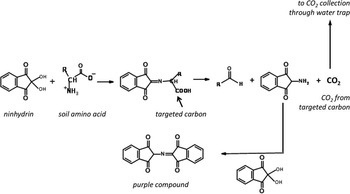
Our chemical procedure derives from the Tisnérat-Laborde et al. (Reference Tisnérat-Laborde, Valladas, Kaltnecker and Arnold2003) protocol developed for bone 14C dating specifically targeting amino acids that constitute bone proteins (collagen). The protocol uses ninhydrin and focuses on the carbon of the carboxyl group in the alpha position of the amine group of organic compounds. The method allows the specific cleavage of the C-C bound caused by the ninhydrin and the subsequent release of CO2 made of the amino acid carboxyl carbon (Figure 1). As the aim of the present study was to target soil amino acid pools, it was quite straightforward to adapt this protocol to soil amino acids.

Figure 1 Principle of the ninhydrin reaction
Soil samples of 3–4 g were introduced into centrifuge tubes. To free the amino acids, peptide hydrolysis was conducted with hot acid (6N HCl at 100°C overnight). The cooled tubes were then centrifuged and the supernatant transferred into a glass flask. The residue was washed twice with 0.1N HCl, the tubes were centrifuged, and the supernatant was transferred to the glass flask. The amino acid solution was evaporated to dryness by means of a rotary evaporator (45°C). The residue was redissolved twice in 2 mL of pure water, transferred to a specific ninhydrin reactor, and evaporated until dryness under nitrogen. The ninhydrin reactor was then connected to the vacuum line and heated to 90°C with heating cords. Then, 2 mL of ninhydrin solution [50 mg of ninhydrin in 2 mL sodium citrate buffer (pH=4.8)] were injected through a septum onto the dry amino acid residue. The targeted carboxyl carbon was released (Figure 1). To extract it from the ninhydrin reactor without flushing the line with the liquid solution, the reaction residue was frozen before the connection to the remaining part of the line. The evolved CO2 was then dried by passing through –80°C dry-ice/ethanol traps, measured, and collected in a flame-sealed Pyrex tube. Glassware was acid-washed and combusted at 450°C overnight prior to use.
The CO2 tubes were shipped to the NSF-AMS Arizona laboratory, Tucson, USA, for 13C and 14C activity measurements. There, the CO2 was transferred, by cracking, into a glass vessel connected to a VG Optima isotope ratio-mass spectrometer for 13C measurement. The remaining CO2 was converted into graphite over iron at 575°C in the presence of ZnO at 425°C (Jull et al. Reference Jull, Burr, Beck, Donahue, Biddulph, Hatheway, Lange and McHargue2003, Reference Jull, Burr, McHargue, Lange, Lifton, Beck, Donahue and Lal2004). Results are expressed in F14C as recommended by Reimer et al. (Reference Reimer, Brown and Reimer2004) to better compare the modern and older samples (Table 1). The carbon concentration of the samples and δ13C and 14C results obtained on the carbon in α-amine position are presented in Table 1. They are complemented by the amount of carbon that was extracted from the soil by the ninhydrin reaction.
Table 1 Experimental results. For the four plots analyzed, the following data are reported: the time elapsed since the C3 to C4 conversion, the sampling year, F14C of atmospheric CO2 in the sampling year (yearly average derived from Hua et al. Reference Hua, Barbetti and Rakowski2013), bulk soil carbon concentration (mean value and standard deviation within three parcels), bulk carbon δ13C, and carboxyl carbon characterization. For the latter, the following data are reported: the mass of soil for the ninhydrin extraction, the mass of exhausted carboxyl carbon, GifA- (chemistry) and AA- (physical measurement) identification numbers, carboxyl carbon δ13C, and F14C. As the 15-yr plot was sampled in 2008, the equivalent F14C in 2002 is also evaluated (error propagation is also reported).

* Typical analytical error ±0.2‰.
Processing Data
The time course of the F14C of atmospheric CO2 during the experiment was obtained from the database for the Northern Hemisphere (NH1) in Hua et al. (Reference Hua, Barbetti and Rakowski2013). The F14C of atmospheric CO2 in the Northern Hemisphere (NH1) decreased from 1.0813±0.0018 in 2002 (individual measurement standard deviation of 0.0030) to 1.0552±0.0048 in 2008 (individual measurement standard deviation of 0.0040) (yearly averages derived from Hua et al. Reference Hua, Barbetti and Rakowski2013), i.e. a 0.0261±0.002 F14C decrease. We applied the same shift to the 2008 sample to obtain a value equivalent to what it would have had if a soil with 15 yr of C4 plants had been sampled in 2002. The propagation of uncertainties yielded 1.023±0.015 for a 2002 equivalent F14C of the 15-yr sample (Table 1).
The proportion of new C (derived from C4 crops) in any fraction was calculated from 13C using the following equation (Balesdent and Mariotti Reference Balesdent and Mariotti1996), which is not biased by 13C fractionation associated with carbon decay in the soil:
 $$X\,{\equals}\,(\rdelta _{{\rm sample}} {\minus}\,\rdelta _{{\rm reference}} )/(\rDelta \,\rm \rdelta _{{vegetation}} )$$
$$X\,{\equals}\,(\rdelta _{{\rm sample}} {\minus}\,\rdelta _{{\rm reference}} )/(\rDelta \,\rm \rdelta _{{vegetation}} )$$
where δsample is the δ13C of the studied material (bulk soil C or carboxyl C), δreference is the corresponding δ13C in the reference wheat soil, and ∆δvegetation is the difference between new vegetation δ13C (mean value of maize plant root and shoot matter –12.2‰) and reference vegetation δ13C (mean value of wheat root and shoot matter –28.2‰), ∆δvegetation=+16‰.
RESULTS
Table 1 presents the yields of the ninhydrin reaction. We obtained less than 260 µg of carbon in all cases and even down to 50 µg in one case, for an aliquot of 3.2–3.9 g of soil. The approximate concentration of protein carbon was 0.6 mg C/g soil and that of carboxyl carbon was ~0.2 mg C/g protein (the carboxyl carbon is an average 4.8 carbons per amino acid molecule). Our yield was therefore probably in the range 15–60%. Part of the loss could be due to deamination during HCl hydrolysis and not to the ninhydrin reaction alone.
The δ13C of carboxyl carbon increased from –42.6‰ to –30.2‰ with the years of C4 plants, whereas the δ13C of bulk soil carbon increased from –26.0 to –24.1‰ (Figure 2). The carboxyl carbon δ13C was much lower than that of bulk δ13C. These findings are similar to unpublished data we previously obtained at LSCE on bone proteins through the ninhydrin treatment that yielded for carboxyl carbon a δ13C value of –32 to –35‰ for a typical bulk bone δ13C of about –19‰ to –20‰. Such a depleted δ13C for carboxyl carbon contradicts the widely published data showing enriched carboxyl carbon (Keeling et al. Reference Keeling, Nelson and Slessor1999; Marsh et al. Reference Marsh, Mulvaney and Sims2003; Apostel et al. Reference Apostel, Dippold, Glaser and Kuzyakov2013). On the other hand, Savidge and Blair (Reference Savidge and Blair2005) also highlighted a depleted isotopic composition for carboxyl carbon with respect to the rest of the molecule (glutamate) by –5 to –13‰. They explained the discrepancy between published studies by different synthesis pathways involving different types of organisms, with carboxyl carbon being strongly depleted in the case of heterotrophic bacteria. Our study might fall within this context, but even so, this cannot explain the whole range of the isotopic depletion. Our protocol might also be the cause of additional isotopic fractionation. The frozen step, used to avoid flushing the line with liquid, is in all likelihood responsible for this fractionation. Likewise, the CO2 gas transport within the vacuum lines including the cracking step for such low amounts of CO2 can be responsible for additional isotopic fractionation. Further work needs to be done to better constrain the physical part of this poorly understood isotopic fractionation.

Figure 2 Proportion of renewed carbon versus the time elapsed since the vegetation conversion: closed circles for carboxyl carbon, open squares for bulk soil carbon. Uncertainty ranges at the bulk scale were calculated based on the three parcels standard deviation and 0.5‰ of deviation for plant δ13C estimates. Uncertainty ranges at the carboxyl carbon scale were calculated based on a standard deviation of 1.5‰ on carboxyl carbon δ13C to take into account the potential nonreproducibility of the ninhydrin fractionation effect.
Nevertheless, as the isotopic fractionation coefficient between bulk and carboxyl carbon was approximatively the same across our four experiments, the relative variation in δ13C remains informative, keeping in mind the high error margin. The 14C activity (F14C) of the carboxyl carbon fraction ranged between 0.932±0.010 and 0.997±0.009 in the maize fields, and 1.035±0.007 for the reference plot (Figure 3). This is less than the F14C of atmospheric CO2 (vegetation carbon) during the experiment (Table 1), indicating the contribution of old carbon.

Figure 3 14C activity of the total carboxyl carbon fraction (closed circle) and of the old carboxyl carbon fraction (open gray diamond). The open circle is for the 2002 equivalent of the 15-yr maize F14C sampled in 2008. Uncertainty ranges of the total carboxyl carbon fraction are smaller than the symbol size. Uncertainty ranges of the F14C estimate of old fraction were calculated by propagating the uncertainty of the renewal rate estimate.
DISCUSSION
Soil amino acid can be split into two pools: a very rapid pool with a mean residence time of about 2 yr that accounts for about three-quarters of the total amino acids and a much more stable older pool for the remaining quarter. Considering an identical shift of +16‰ between C4-derived carboxyl C and C3-derived carboxyl C, the expected C4 carboxyl carbon δ13C pole should tend towards –26.6‰. This means that with the –30.2‰ reached after 15 yr of C4 plants, 78% of the amino fraction pool has been renewed (Equation 1]. Most of the renewal was already reached after 9 yr. This has to be compared with the ~15% of renewed carbon in the bulk pool. The amino fraction turnover is much faster than that of the soil average carbon. Our study provides quantitative information on the amino fraction turnover rate: a plateau is reached between 9 and 15 yr, leading to a differentiation of at least two pools of carboxyl carbon, the youngest one being totally renewed after 9 yr. This means that it takes less than 9 yr to get a 75–80% renewal of the amino fraction in this Cambisol. The mean age of the carbon in this fast component of the amino-C pool is ~2 yr (Figure 1). As established by Dippold et al. (Reference Dippold, Biryukov and Kuzyakov2014), the carboxyl carbon has a more rapid turnover than the remaining part of the amino acid molecule. It remains only a few days in the citric cycle, whereas the remainder of the molecule stays longer in the citric cycle. It is thus likely than the mean age of the remainder of the molecule is higher than that of the carboxyl carbon. The plateau value, which is reached between 9 and 15 yr, indicates that the remaining 20–25% amino acid C is much more stable and older.
The old amino acid pool shows a mean age of several thousands of years. As we estimated a mean age of 2 yr for the young amino acid pool, it is possible to approximate its 14C activity (F14Cyoung) by atmospheric 14C activity 2 yr before the sampling: F14Cyoung=1.093 for the 2002 sampling and F14Cyoung=1.064 for the 2008 sampling. Furthermore, we estimated that the young amino acid pool accounts for 77% of the total amino acid pool. This means that the remaining old amino acid pool shows a much lower 14C activity, of F14C=0.4, 3 yr after the vegetation conversion to 0.8 before the conversion and after the 15 yr of maize. We thus conclude that the F14C of “old” carboxyl C shows a mean age of several thousands of years.
In greater detail, the amino 14C activity depended significantly on the plot (Table 1): it was high in the wheat reference field, drastically decreased just after the vegetation conversion, and increased thereafter. In view of the rapid turnover of this fraction, a rapid evolution of its amount can be suspected as well, e.g. associated with the N balance of the crop, which depends on inorganic fertilization, inorganic N loss, and plant nitrogen recycling by crop residues. A more precise quantification would be required to investigate this hypothesis. Other hypotheses can be put forward to explain the presence of old amino acid: (1) newly bio-assimilated carbon derived from a 14C-depleted mixture of compounds; (2) the carboxyl carbon pool lost 14C-enriched compounds (carbon from the 1960s); and (3) both (1) and (2).
According to the model proposed by Kleber et al. (Reference Kleber, Sollins and Sutton2007), soil organic matter organization can be represented as a multilayer structure combining organomineral interaction and heterogeneous interactions between organic materials. This conceptual model leads to specific and potentially long-term preservation of proteins in the “contact zone,” i.e. the zone the closest to the mineral and consequently the least accessible for microorganisms. The compactness of the layers (contact zone and subsequent zones) relates to external parameters such as physical properties (e.g. pH, redox), soil properties (e.g. clay content, clay type), and soil-plant interactions. A change in vegetation may have implied changes in one or more of the above properties and as a result, in a decrease of the compactness of the multilayer structure of SOM. Vegetation conversion distends organomineral and organo-organo interactions, leading to a higher accessibility of previously protected organic compounds.
In such a context, old soil organic compounds may have been a source of carbon for newly biosynthetized amino fraction constituents. This can be the case for old components previously tightly bound to the mineral structure or in the Closeaux context, which is known to include lignite and domestic combustion residues, bound to activated charcoals (Dippold et al. Reference Dippold, Biryukov and Kuzyakov2014). This use of old organic compounds might also be related to the priming effect (Fontaine et al. Reference Fontaine, Barot, Barré, Bdioui, Mary and Rumpel2007). New compounds deriving from plant residues brought fresh energy to microorganisms. As a result of this fertilization, microorganisms became able to use old neglected compounds as a source of carbon, assimilating 14C-depleted carbon within their structure. Both ideas illustrate assumption 1.
Likewise, some amino compounds a few decades old may have been mineralized during the field experiment, leading to a loss in enriched 14C compounds (more strongly imprinted by the bomb peak) and thus to a decrease in the 14C activity of the average amino fraction. This illustrates assumption 2.
A more effective citric cycle (Dippold et al. Reference Dippold, Biryukov and Kuzyakov2014) can be invoked to explain the concomitance of the two phenomena: assimilation of old compounds and mineralization of compounds a few decades old. This effect fades as the soil returns to a steady state. Input of modern 14C compounds matches the increase in the 14C activity of carboxyl carbon. Our study thus highlights and quantifies the priming effect on a temperate cultivated soil.
CONCLUSION
The present study provides the first data of the turnover of the amino acid fraction of soil organic matter, based on natural isotope abundance of the carbon in alpha position of the amino group. Our results suggest that the carbon of the carboxyl group belongs to two different compartments, each with its own turnover dynamics. 14C changes over time, after a cultivation change from C3 to C4 crops, point to a priming effect in which older compounds become available to microorganisms.
ACKNOWLEDGMENTS
Samples were taken within the French ANR-DynaMOS project (ANR-07-BLAN-0222). Analysis and interpretation took place within the ANR 14-CE01-004 DedyCAS project. This study fits with the LabEx BASC thematics. We are particularly indebted to the friendly contribution of the INRA-Agro-Paristech team in charge of the long-term experiment SolFit in Versailles: Claire Chenu, scientific manager and Jean-Pierre Pétraud, operational manager. The content of this paper greatly benefits from the comments of three anonymous reviewers. This is an LSCE contribution n°5995.