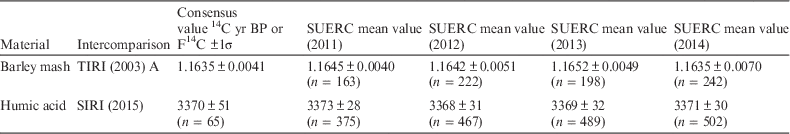INTRODUCTION
The Scottish Universities Environmental Research Centre (SUERC) Radiocarbon Dating Laboratory was first established in the 1960s at the University of Glasgow and the first significant publication arising from the laboratory’s research was published in Nature (Walton and Baxter Reference Walton and Baxter1968). In 1986, the laboratory was relocated to the Scottish Universities Research and Reactor Centre (SURRC, subsequently renamed SUERC) in East Kilbride, where radiometric analysis by liquid scintillation spectrometry was undertaken until the implementation of AMS 14C dating in 1998. The AMS graphite preparation system was set up with the help and guidance of the NSF-Arizona AMS Facility at the University of Arizona, where the prepared samples and standards were initially sent for 14C measurement. Following the installation of a National Electrostatics Corporation 5MV tandem AMS instrument at SUERC in 2003, the radiocarbon laboratory, in collaboration with AMS laboratory staff, have been undertaking 14C AMS analyses (Xu et al. Reference Xu, Anderson, Bryant, Cook, Dougans, Freeman, Naysmith, Schnabel and Scott2004; Naysmith et al. Reference Naysmith, Cook, Freeman, Scott, Anderson, Xu, Dunbar, Muir, Dougans, Wilcken, Schnabel, Russell, Ascough and Maden2010). This capability was subsequently increased in 2007 with the establishment of a National Electrostatics Corporation 250kV single-stage AMS instrument. To date, approximately 27,000 14C ages have been measured by the AMS facility and, currently, around 3500–4000 unknown-age samples are measured annually. A range of sample types is processed in the laboratory from the disciplines of archaeology, environmental science, forensic science, ecology, and the petrochemical (biofuel) industry. In this paper, we provide detailed laboratory procedures for sample submission, logging of samples into our custom-designed database (Tripney et al. Reference Tripney, Naysmith and Cook2014), pretreatment of a range of organic and inorganic sample types, sample combustion/hydrolysis and CO2 graphitization, AMS analysis and data reduction, associated stable isotope measurements, data handling processes, and Bayesian analysis.
SAMPLE SUBMISSION
Sample submission forms and dating certificates have been maintained for all radiometric samples measured at the SUERC Radiocarbon Laboratory since 1965. From 1998, with the introduction of AMS, an additional electronic record of all AMS samples was introduced in the form of an Excel spreadsheet. At the point of receiving a sample into the laboratory, each sample is designated a specific laboratory number beginning with the prefix GU- to signify processing at the SUERC Radiocarbon Laboratory. This is the defined number for the sample during the course of all processes within the laboratory and for tracing all sample data. However, at the AMS measurement stage, the sample is given an analysis code with the prefix SUERC- to signify measurement at the SUERC AMS facility. The recommended pretreatment scheme for the sample is also selected and the requested turnaround time defined. Requests for ancillary stable isotope analyses are also added, if necessary. However, with the large volume of samples processed in the laboratory, the data handling within this spreadsheet-based system became impractical and led to the development of a custom-designed electronic database in 2011 (Tripney et al. Reference Tripney, Naysmith and Cook2014). Each sample submitter completes a simple Excel spreadsheet with both their affiliation and sample details (site, sample type, species dated, sample ID, and context ID using a single row per sample). A hard copy of the spreadsheet should accompany the samples while an electronic copy is emailed to the laboratory, where it is loaded into the database together with the laboratory’s selection of an appropriate pretreatment scheme. The benefit of this system is that it eliminates transcription errors on the part of laboratory staff. This triggers the printing of “laboratory forms” that contain the laboratory code (GU-) of the sample, the pretreatment scheme to be used, the sample deadline, and a number of boxes for sample weight, pretreated sample weight, mass of sample used in the analysis, yields at each stage of the process, and a comment box for any notes on anything unusual such as sample appearance, pretreatment losses, etc. This form tracks the progress of the sample through each stage of the dating process, while the member of staff working on the sample is responsible for entering the data into the database.
SAMPLE PREPARATION AND PRETREATMENT
As a prerequisite, all pretreatment steps are carried out in a fume hood and all glass vessels and implements used are precleaned by washing in a 5% solution of Decon® 90 (surface active decontamination solution) and rinsing with 0.1M HCl followed by Milli-Q® grade ultrapure, reverse osmosis water. This quality of water is used throughout all pretreatments for rinsing and preparation of solutions, and the carbon content is checked routinely. Following this cleaning, the glassware and implements used in pretreatment and graphitization are heated at 500°C and allowed to cool overnight. All chemical reagents used are analytical grade or better.
Bone
The laboratory practices a modification of the Longin (Reference Longin1971) procedure for the routine extraction of collagen from bone samples. Advances in pretreatment such as ultrafiltration (Bronk Ramsey et al. Reference Bronk Ramsey, Higham, Bowles and Hedges2004) have been used to improve the quality of bone collagen extracted by the removal of smaller molecular weight compounds (<30 kDa). This fraction is assumed to be either degraded collagen or exogenous material. The introduction of the ultrafiltration method has been extensively trialled and is now routinely undertaken in the laboratory when requested. It is well accepted that the ultrafilters may contain residual contaminant carbon from the humectant present in the filters, if they are imperfectly cleaned prior to use. In order to monitor the amount and potential “age” of this humectant, the filtrate solution from one cleaned ultrafilter is retained and measured in each batch of bone samples. The residue is combusted, graphitized, and measured by AMS. Preliminary measurements have given an average value of F14C=1.0253±0.0027, which is in agreement with those values determined at ORAU (Brock et al. Reference Brock, Higham and Bronk Ramsey2013). Both the modified Longin method and the ultrafiltration method are detailed below. As a matter of procedure, δ13C, δ15N, and the C/N ratio are measured on subsamples of the prepared collagen to assess its suitability for dating. In line with DeNiro and Hastorf (Reference DeNiro and Hastorf1985), any samples with a C/N ratio outside the range 2.9–3.6 would be deemed to represent collagen that has undergone postdepositional alteration and would not be considered for 14C analysis. δ34S can also be determined on a separate collagen subsample if requested.
Modified Longin Method
A subsample of the bone (1–2 g) is cut and the bone surfaces cleaned to remove any adhering soil and contaminant material using a Dremel multitool. The bone fragment is weighed and the weight recorded. After close inspection to establish that ink, varnish, or other contaminants associated with museum curation are not present, the bone is added to 100 mL of 1M HCl and allowed to react for a minimum of 24 hr to dissolve the apatite, after which, the remaining bone material should appear “jelly like.” The excess acid is decanted off, the sample is rinsed in ultrapure water, and a further 100 mL of ultrapure water are added and the sample heated in a sand bath at approximately 80°C for 3 hr. When the bone is completely solubilized, the solution is allowed to cool and then filtered using GF/A filter (Whatman) paper. The collagen solution is dried down to <20 mL and transferred to a weighed vial. The vial is then transferred to a freeze-drier until all the solution is completely removed and the collagen is left as a white/off-white crystalline powder.
Modified Ultrafiltration Method
The procedure is the same as the modified Longin (Reference Longin1971) method described in the previous section, up to the point of drying down the collagen solution. At this point, the collagen solution is transferred to a precleaned Vivaspin 20TM filter, centrifuged at 4500 rpm for 30 min, and transferred to a preweighed 20-mL vial. The sample is then freeze-dried to produce a white/off-white crystalline powder. [N.B. Thorough cleaning of the Vivaspin 20 filters is absolutely essential since they contain a humectant. Those that have been examined recently have a modern F14C signature as described by Brock et al. (Reference Brock, Higham and Bronk Ramsey2013). To remove this humectant, the filters are rinsed several times with ultrapure water, sonicated for 30 min, and finally rinsed again for immediate use (Bronk Ramsey et al. Reference Bronk Ramsey, Higham, Bowles and Hedges2004; Brock et al. Reference Brock, Bronk Ramsey and Higham2007, Reference Brock, Higham and Bronk Ramsey2013).]
There are important criteria that must be met before proceeding with the dating of any collagen sample. At the pretreatment stage, the total collagen yield is an important indicator of the preservation state of the bone and its suitability for dating. Initial bone weight and collagen weight are recorded in the database, which is set to flag up any sample with a collagen content of less than 5% of the starting weight of bone. Between 1 and 5% it is a judgement call by one of the senior staff on whether or not the sample is dated. If the amount of collagen extracted from the original sample is <1% of the starting weight of bone material, the sample is rejected. Second, the C/N atomic weight ratio of the collagen [measured separately by continuous-flow isotope ratio mass spectrometry (CF-IRMS)] should be in the range of 2.9–3.6 (DeNiro and Hastorf Reference DeNiro and Hastorf1985; van Klinken Reference van Klinken1999). Both of these criteria must be met before proceeding with the analysis as values outside the ranges may indicate significant contamination or degradation of the bone. It should also be noted that when impurities such as humic substances are present, a NaOH step may be added before collagen extraction, but the laboratory will not routinely include this alkali wash as pretreatment losses are typically much higher when a NaOH step is included.
Charcoal and Carbonized Grain
Charcoal and carbonized grain samples are the most common sample types currently pretreated in the laboratory (~30% of the total sample throughput). For most samples, a simple acid-base-acid (ABA) pretreatment is sufficient to ensure complete removal of non-sample-derived carbon. The minimum weight for an analysis is 0.0030 g (3 mg) with a preferred weight of around 0.1000 g since a significant loss in weight can occur during the pretreatment steps. The laboratory only undertakes single-entity dating unless otherwise requested. The reasoning was put forward by Ashmore (Reference Ashmore1999) for our work with Historic Scotland and was immediately extended to all work unless there is a compelling reason to do otherwise. A single fragment of charcoal or a single carbonized grain is selected and weighed. Some 100 mL of 0.5M HCl are added to the sample, which is heated on a hotplate for 2 hr at ~80°C. This step will both dissociate any carbonate material present and remove acid-soluble impurities. The solution is allowed to cool and then discarded. The sample is then rinsed with ultrapure water before 100 mL of 0.5M NaOH are added and the sample heated on a hotplate to ~80°C for a further 2 hr. This step removes alkali-soluble contaminants, such as humic acids (HA), and if necessary is repeated until the NaOH solution is clear. The solution is then decanted, the sample rinsed with water before the addition of 100 mL of 0.5M HCl, followed by heating to 80°C for 2 hr. The additional acid step is required to neutralize excess NaOH. The sample is then rinsed with ultrapure water and dried overnight. If the sample is small (<3.0 mg), the total sample is transferred directly into a precombusted, weighed quartz insert, which is placed directly in a quartz combustion tube.
Several studies (e.g. Higham Reference Higham2011) have reported improvements in ages obtained from older charcoal by adopting the acid-base-oxidation, stepped combustion technique first developed by Bird et al. (Reference Bird, Moyo, Veenendaal, Lloyd and Frost1999). However, since all the charcoal samples analyzed in the laboratory are <2 half-lives in age (and most are <1 half-life), we have found that this additional pretreatment step is unnecessary.
Pottery Sherds and Carbon Residue
In the case of the pretreatment of carbon residues, the samples arrive in the laboratory either on the surface of a pottery sherd or as a separated carbon residue. If the sample is attached to a pottery sherd, the carbon residue must be removed with care using the flat edge of a spatula or scalpel onto aluminium foil. The carbon residue must be gently pushed from the surface and not scraped, as the latter can result in the inclusion of mineral material from the sherd. This mineral material can potentially include carbon from organic coatings on the clay minerals that were not totally removed by the initial firing. This carbon can be significantly older than the date of firing. The carbon residue is transferred to a small glass beaker, weighed, and then pretreated in the same manner as carried out for charcoal and carbonized cereal grains.
Cremated Bone (Groningen Method)
Bone samples submitted for analysis as cremated bone must be gray and preferably almost white in color, indicating that they have been burned at >600°C (Lanting et al. Reference Lanting, Aerts-Bijma and van der Plicht2001; Van Strydonck et al. Reference Van Strydonck, Boudin and De Mulder2009). If the bone has simply been charred and is brown in color, it should not be analyzed as the carbon contained within the apatite structure will not be fixed and exchange could have occurred (Van Strydonck et al. Reference Van Strydonck, Boudin and De Mulder2009). The percentage carbon in a cremated bone is very low, normally in the range 0.1–0.5%; therefore, the minimum weight required will be at least 1 g, with an optimum weight in excess of 5 g. A single bone fragment is selected, weighed, and recorded, unless the cremated bone is totally fragmented (<5 mm diameter), in which case, more than a single fragment is used, provided the fragments can be demonstrated to come from a single cremation. The bone surfaces are initially cleaned with water to roughly remove any adhering soil and then placed in an ultrasonic bath for 10 min. Following this initial cleaning, the bone is soaked in 100 mL of 1.5% sodium hypochlorite solution for 48 hr to oxidize any residual organic material incorporated from the burial environment. The sodium hypochlorite solution is then decanted and the bone fragments rinsed thoroughly with ultrapure water (10×250 mL). A further 250 mL of water are added to the bone and the sample is allowed to stand at room temperature for 1 hr before proceeding. The water is then decanted and 100 mL of 1M acetic acid are added to remove the more soluble carbonate ions as well as the less crystalline and more soluble fractions of the bone apatite. After 24 hr, the acetic acid is decanted and the bone fragments repeatedly rinsed with cold water (10×250 mL). A further 250 mL of ultrapure water are added to the bone and left for 1 hr. The water is then decanted and the sample oven-dried at 40°C for 24–48 hr. The dried sample is then powdered using a pestle and mortar and transferred to a labeled glass vial to await analysis.
Contemporary Tooth Enamel
The method described is for the 14C analysis of contemporary tooth enamel, which can be used to estimate year of birth (Spalding et al. Reference Spalding, Buchholz, Bergman, Druid and Frisén2005; Cook et al. Reference Cook, Dunbar, Black and Xu2006). The laboratory does not presently recommend the dating of archaeological tooth enamel. We have dated tooth enamel from a number of sites and produced entirely reasonable results, but age anomalies between collagen (from both tooth and bone) and tooth enamel have been obtained in a recent study.
Tooth enamel has a similar structure to that of cremated bone, both comprising biological apatite (calcium hydroxyapatite), which has the formula Ca10(PO4)6(OH)2. Tooth enamel comprises almost 96% mineral material and the percentage carbon is very low, normally in the range 0.1–0.8%. The required minimum weight of tooth is 0.5 g. This is placed in a beaker to which 100 mL of ultrapure water are added and the beaker placed in a sonic bath for 5 min to remove any adhering soft tissue. The sample is then rinsed and the procedure repeated if necessary. Using a Dremel multitool, the crown is removed, cut in half lengthwise, and the internal dentine material removed as far as possible from each section using a dissecting needle. The crown is placed in 10M NaOH solution, heated to ~80°C for 8 hr and allowed to cool. The dentine is again scraped from the enamel using a dissecting needle and the whole step is repeated until all the dentine has been removed. The sample is repeatedly rinsed with 0.5M HCl to remove all NaOH and finally rinsed with ultrapure water. The isolated enamel is then oven-dried overnight and transferred to a labeled glass vial to await analysis.
Shell
The shell surface is mechanically cleaned to remove adhering detrital material and placed in a beaker with 100 mL of water. This is placed in an ultrasonic bath for 10 min to remove any remaining material. The sample is then rinsed with ultrapure water and the procedure repeated if necessary. The total weight of sample is recorded. The optimum weight is >0.1 g. Approximately 20% of the shell surface is removed by appropriate addition of 1M HCl. This eliminates potential surface and edge contaminant CaCO3. The sample is rinsed with ultrapure water and oven-dried overnight.
Wood
There are two categories of wood sample that the laboratory receives for 14C measurement. The first is from environmental contexts or from archaeological sites. Most of these require a relatively mild pretreatment as they are often delicate in nature and the main contaminants are humic substances. The second type of wood sample is that taken from built structures or samples for wiggle-matching. Those from built structures can differ in requirement as they are more prone to potential sources of anthropogenic contamination, and as such these are more thoroughly pretreated by isolating the alpha-cellulose fraction, as described below, to ensure efficient elimination of all possible sources of contaminant carbon. The alpha-cellulose component is the most reliable fraction for determining the 14C at the time of growth and as such is the established method used for wiggle-matching.
Wood samples represent approximately 6.5% of the total number of samples received by the laboratory and only a small percentage of these require the full alpha-cellulose procedure. Again, for most wood samples, a simple acid-base-acid (ABA) pretreatment, as described earlier, is sufficient to ensure complete removal of non-sample-derived carbon. The minimum weight required is 0.01 g (10 mg) with an optimum weight of >0.1 g, since significant loss of weight is expected during the pretreatment step, especially in waterlogged samples. The subsample taken is dependent on the overall sample size and ring structure. Where a large section with many rings has been submitted, a small section of the outer few rings is removed and pretreated. If the sample appears sufficiently robust, a cold bleaching step for 24 hr (50 mL of 0.2M sodium hypochlorite) may be used to remove the lignin fraction.
Alpha-cellulose preparation requires sequential solvent extraction steps to remove waxes, oils, and resins as well as other potential organic contaminants such as varnish present in the wood. The laboratory employs a modification of the Hoper et al. (Reference Hoper, McCormac, Hogg, Higham and Head1998) method. For the three-step Soxhlet extraction process, the wood sample is weighed, cut into thin slivers, and placed in a glass-fiber extraction thimble plugged with glass wool. An appropriate volume of chloroform/ethanol CHCl3/C2H5OH (2:1 by volume) is added and the sample refluxed for 8 hr. The solvent is allowed to cool and discarded. This process is repeated using ethanol only and finally with distilled water. The sample is dried overnight at <30°C.
The addition of a cold bleaching step for 24 hr [50 mL of 0.2M sodium hypochlorite (NaClO)] will remove the lignin fraction. If the wood sample is small or fragile, this concentration should be reduced and the length of time in the bleach solution adjusted. After the wood is white in color, the excess solution is decanted off and the sample rinsed with water. Some 100 mL of 0.5M HCl are added to the sample, which is heated for 2 hr at 80°C to ensure the complete removal of excess sodium hypochlorite solution. The solution is allowed to cool and then discarded while the sample is rinsed with water before 100 mL of 0.5M NaOH are added and the sample heated for 2 hr. This alkali step breaks down hemicellulose and is repeated until the solution appears transparent. The sample is then rinsed with water before the addition of 100 mL of 0.5M HCl. Again, it is heated for 2 hr to neutralize any remaining alkali. Finally, the sample is rinsed with water and oven-dried at <30°C.
Peat, Soil, and Organic Sediments
In the case of peat, the most commonly dated fractions are macrofossils that can be identified as fragments of the parent vegetation forming the peat, or in cases where the peat is so well humified that these cannot be recovered, we typically date the humic acid fraction. Where possible, we will also date the humin fraction as agreement between these two fractions provides added confidence in the results, if requested.
We recommend caution when dating accumulating sediments as these can be secondary deposits in which the carbon may be significantly older than expected. Similarly, we do not recommend dating soils to produce an age although a great deal of information can be obtained on organic matter turnover from dating different fractions of this material, especially surface soils containing a nuclear weapons testing signal (e.g. Harkness et al. Reference Harkness, Harrison and Bacon1986). Buried soils are somewhat different and where identifiable macromaterial deriving from the buried vegetation layer can be identified, we would analyze this. The chemistry of humic substances is incredibly complex and is still relatively poorly understood. Humic substances can be divided into three main operationally defined fractions: fulvic acid is acid and alkali soluble, humic acid is acid insoluble/alkali soluble, and humin is insoluble in both acid and alkali. Humic substances are a product of microbial action on dead organic matter and humic acid is a major component of humic substances. Humic acid does not have a defined structure but is a complex combination of many different organic acids with numerous and varied functional groups. The specific properties and structure of a humic acid fraction are dependent on many different factors, including the method of extraction. This fraction is preferred for dating because it is acid insoluble in a typically acid pH environment and is less likely to be influenced by groundwater movement than the fulvic fraction. In contrast, the humin fraction will naturally contain all material resilient to biodegradation and can include living roots and in-washed material from elsewhere in the depositional environment.
If macrofossils are the preferred fraction for dating, we prefer that these are selected by an expert in this field. The macrofossils are prepared using a slightly modified version of the ABA method used for charcoal samples.
Humic Acid Fraction
For the humic acid extraction, the minimum weight required is 1 g, but if possible, we prefer a major part of the depth increment of the peat sample that has been recovered. Peat does not form in horizontal layers, and so a large sample produces an averaging of the age from a particular depth increment. Some 250–500 mL of 0.5M HCl (or a volume proportional to the size of sample) are added to the sample and heated to 80°C for 2 hr. The acid step primarily removes the acid-soluble fulvic acid component. This extract is discarded and 100 mL of 0.5M NaOH are added to the sample, which is again heated to 80°C for 2 hr. The sample is allowed to cool and the solids to settle before filtering the extract into a separate beaker. This NaOH step is repeated to ensure all the humic acid is extracted. Excess 4M HCl is added to this NaOH solution, which is heated for ~1 hr to precipitate the humic acid fraction. The resulting solution and the precipitate are allowed to cool and the solids settle. The solid fraction (alkali soluble and acid insoluble) is the humic acid. The solids are transferred to a centrifuge tube and spun and rinsed several times with water to remove all trace of fulvic acid and residual salt from the precipitation step. The solid residue is then transferred to a freeze-drier until all the solution is removed and the humic acid, in the form of a crystalline powder, remains.
All residual material, after the HCl and NaOH extraction steps, is defined as the humin (insoluble in acid and alkali). If this is required for dating, 100 mL of 0.5M HCl are added to the humin material and heated to 80°C for 1 hr to neutralize any remaining NaOH. The remaining material is then filtered and freeze-dried and ready for analysis.
Plant Macrofossil
Plant macrofossil samples are the most fragile sample type pretreated in the laboratory and a simple ABA pretreatment is sufficient to ensure complete removal of non-sample-derived carbon. The minimum weight for an analysis is 0.0030 g (3 mg); however, a significant loss in weight can occur during the pretreatment steps and so we would advise that around 10 mg are submitted. The laboratory only undertakes single-entity dating unless otherwise requested, and as such, the method must be amended accordingly. Some 25 mL of 0.1M HCl are added to the sample, which is heated on a hotplate for 2 hr at 80°C. The supernatant is allowed to cool and discarded and the sample rinsed with ultrapure water before 25 mL of 0.1M NaOH are added and the sample heated for a further 2 hr. The solution is then decanted, the sample rinsed with water before the addition of 25 mL of 0.1M HCl, followed by heating to 80°C for 2 hr. The sample is then rinsed with water and dried overnight. If the sample is small (<3.0 mg), the total sample is transferred directly into a precombusted, weighed quartz insert, which is placed directly in a quartz combustion tube and dried overnight at ~30°C.
Textiles
Textiles are very delicate in nature and require adaptation of a defined pretreatment method dependent on each sample. Sequential solvent extraction using a Soxhlet apparatus will remove dyes or organic treatments applied to the fabric. For the Soxhlet extraction process, the fabric sample is weighed, cut into two, with half placed in a glass-fiber extraction thimble plugged with glass wool. The same sequences of steps that are employed for alpha-cellulose extraction are used. Following the Soxhlet extraction, the sample is then subjected to an ABA pretreatment scheme similar to that for carbonized grain samples.
Forensic Materials
The laboratory undertakes the analysis of several different types of samples each year to assist in police investigations. These include human remains, rhinoceros horn, elephant tusk (ivory), and animal fur (from endangered species). The pretreatment of these is based on those detailed above and, routinely, a combination of several methods is required to purify the sample sufficiently. Specifically, in the case of recent human bone, significant lipid material will be present, which may alter the C/N ratio and δ13C values characteristic of bone collagen. Such samples will require an appropriate solvent extraction and ultrafiltration step if necessary. In addition, the extracted lipid fraction can be useful in confirming the recent 14C value of the degraded tissue (Cook et al. Reference Cook, Ainscough and Dunbar2015). Lipid removal requires Soxhlet extraction. The bone sample is weighed and the surface cleaned to remove any adhering tissue and placed in a preweighed glass-fiber extraction thimble plugged with glass wool. An appropriate volume of chloroform/ethanol CHCl3/C2H5OH 2:1 (v/v) is added and the sample refluxed for 8 hr. The solvent is allowed to cool and retained if further analysis of the residue is required. This process is repeated to ensure complete lipid removal. This is followed by further extraction using ethanol, and finally the sample is rinsed several times over a 2-day period with ultrapure water and dried overnight at ~30°C before proceeding with the collagen extraction detailed in the previous section on bone analysis.
Rhinoceros horn is comprised predominantly of keratin, which is similar to human nail and hair and requires an acid wash only to remove any surface contamination. Conversely, elephant tusk (ivory) is similar in composition to dentine found in human teeth and comprises a collagen matrix with a mineral component; therefore, the collagen fraction is isolated using our modified Longin method.
Animal fur and skin (or leather) also require pretreatments to achieve the complete removal of substances that may have been applied to them postmortem. Where an animal pelt with fur is submitted, we would date the fur as recommended by Geyh (Reference Geyh2001). He has demonstrated that the 14C value of leather (skin) is a complex function of the dates of birth and death and the types of food consumed, making analysis of this material unsuitable for a reliable and precise estimation of the death date using the nuclear-weapons 14C peak. On the other hand, he noted a good fit of the 14C activities of corresponding hair (fur) samples, including most of the carnivores analyzed, to the nuclear-weapons 14C peak (Hua et al. Reference Hua, Barbetti and Rakowski2013) if a bias of 1 year is assumed, where this bias represents the mean age of the food that the animal consumed. The pretreatment for such samples is again by Soxhlet extraction followed by ABA as undertaken for the pretreatment of textiles.
Fuel and Biofuel Samples
The Kyoto Protocol under the United Nations Framework Convention on Climate Change (UNFCCC) is an international treaty that sets binding obligations on industrialized countries to reduce emissions of greenhouse gases. As a result, many developed countries and organizations have agreed to legally binding limitations and reductions in their emissions of greenhouse gases. Several approaches have been employed to reduce the overall CO2 generated, one being the inclusion of a biocomponent in fossil fuel. A method has been developed for the routine measurement of fuel and biofuel in the laboratory as a tool to quantify the percentage biofuel present, in compliance with these guidelines.
All fuel and biofuel samples are stored in a freezer at –20°C prior to their analysis, to minimize the loss of any volatile components. An approximate 5-µL aliquot of the liquid sample is measured into a preheated quartz capillary tube and transferred to a round-bottomed quartz combustion tube containing 0.5 g CuO and 0.1 g silver. The sample is attached to a vacuum sealing rig and immediately cooled to –196°C using liquid nitrogen, ensuring that all volatile fractions are retained. The combustion tube is then evacuated and sealed. It should be noted that the sample must remain frozen under vacuum at all times during the sealing process and additional care must be taken at this point to minimize the loss of volatile components from the sample, which can pose a potential problem during the sealing of the quartz tube. The sample is then combusted overnight, using stepped temperature increments over several hours to a maximum temperature of 850°C for 4 hr.
On completion of all sample pretreatments, the responsible staff member logs the completion date into the database, which records this information in case of any requests for an update on progress
CO2 PRODUCTION, PURIFICATION, AND GRAPHITIZATION
Organic Carbon-Containing Samples
For organic samples, an appropriate weight of material (typically 10–20 mg) is weighed into a clean quartz insert and placed into a precleaned quartz combustion tube containing copper oxide to provide the oxygen for the reaction and silver foil to remove gaseous impurities (Vandeputte et al. Reference Vandeputte, Moens and Dams1996). We regard this as the “gold star” method of generating CO2 from organic samples and one of the reasons for our ability to generate consistently low organic background F14C values.
Inorganic Carbon-Containing Samples
Cremated Bone
Approximately 1 g of the powdered, cremated bone is weighed into the main body of a 250-mL hydrolysis unit. Approximately 50 mL of 15M orthophosphoric acid are measured into the side arm of the unit, which is then transferred to a vacuum line and evacuated for 5–10 min. The tap on the hydrolysis unit is closed and the unit removed from the vacuum line. The phosphoric acid is then added to the cremated bone and left to react in a fume cupboard. After 24 hr, the hydrolysis unit is transferred to a vacuum line where the CO2 produced is isolated, purified by cryogenic pumping, and measured. However, this CO2 is still often sufficiently impure that it does not graphitize. Therefore, an aliquot of the “impure” CO2 is transferred to a quartz combustion tube containing 0.5 g CuO and 0.1 g silver, evacuated, sealed, and furnaced overnight at 850°C. This practice removes impurities commonly produced in the cremated bone hydrolysis step, such as hydrogen sulfide, which prevent the graphitization step from proceeding. A second CO2 aliquot is held as an archive sample if repeat analysis is required.
Tooth Enamel
The enamel is weighed into the main body of a small 100-mL volume hydrolysis unit and 20 mL of 15M orthophosphoric acid are added to the side arm of the unit. The hydrolysis unit is connected to a vacuum line and evacuated for 5–10 min. The tap on the hydrolysis unit is then closed and the unit removed from the vacuum line. The phosphoric acid is added to the enamel and left to react in a fume cupboard for 24 hr. The hydrolysis unit is then reconnected to the vacuum line and the CO2 isolated, cryogenically purified, and finally, the volume produced is measured.
Shell
For CO2 generation, 0.1 g is weighed into a hydrolysis unit (a large, single fragment of shell is normally selected rather than fine material), where a further 20% of the shell is reacted with the appropriate volume of 1M HCl. The hydrolysis unit is assembled with an additional 4 mL of 1M HCl in the side arm. The unit is reattached to the vacuum line, pumped, removed from the line, and the acid is added under vacuum to hydrolyze the remaining carbonate material. The CO2 is then collected, cryogenically purified, and the volume is measured.
Routinely, 3-mL subsamples of CO2 are converted to graphite using the zinc and iron reduction method described by Slota et al. (Reference Slota, Jull, Linick and Toolin1987). Graphite production is monitored using an in-house software program that collects data from the pressure transducer attached to each graphite production unit and calculates the graphite conversion yield from the reduction in CO2 pressure in the unit. The sample must achieve >95% conversion yield for the graphite to be acceptable for dating. When the graphite run is complete, the final yields are sent to the database. The graphite is then pressed into an aluminium cathode for AMS measurement.
Quality Assurance
Preparation and analysis of appropriate quality assurance samples is essential for monitoring the quality of our analyses. Our in-house quality assurance program involving primary, secondary “known-age” and tertiary “in-house” standards is well established. The known-age materials have been used in intercomparison studies between 14C laboratories worldwide and have well-defined consensus age/activity values. The results of all known-age standards are referenced directly to the oxalic acid primary standard (SRM-4990C).
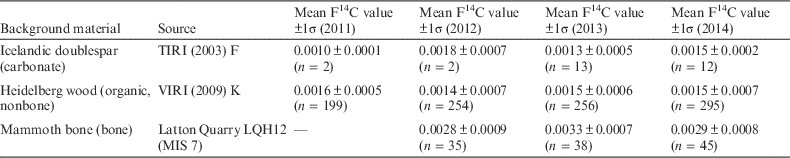
The organic (nonbone) background standard is an interglacial wood used in the Fifth International Radiocarbon Intercomparison (VIRI) (Scott et al. Reference Scott, Cook, Naysmith, Bryant and O’Donnell2007, Reference Scott, Cook and Naysmith2010a, Reference Scott, Cook and Naysmith2010b) (VIRI sample K), provided by Michael Friedrich, University of Hohenheim. (This wood was prepared as described in the section on alpha-cellulose preparation.) When unknown carbonate samples are included in a wheel, a geological-age carbonate in the form of Icelandic doublespar (TIRI sample F) is used to generate CO2 by acid hydrolysis. Carbonate samples make up a very small percentage of the samples analyzed. The background value from the doublespar is indistinguishable from the wood background. For example, in 2014, the mean doublespar background F14C=0.0015±0.0003 compared to 0.0014±0.0006 for the wood. For bone collagen analyses, a mammoth bone originating from a deposit that correlated with Marine Isotope Stage 7 (MIS 7) (Cook et al. Reference Cook, Higham, Naysmith, Brock, Freeman and Bayliss2012) is used to monitor the inherent background value of the bone analysis process. Typically, the background value for bone is somewhat higher than for either the wood or doublespar samples, and this is the subject of a forthcoming paper. Every batch of 134 cathodes will typically contain seven background samples, which are analyzed and used to produce a mean value for the batch. This value represents the background value for the entire process, including laboratory pretreatment, graphitization, and measurement, giving a reliable indication of the achievable age limit.
The secondary archaeological-age standard used since 2010 is a humic acid that was employed in both the VIRI and SIRI (Sixth International Radiocarbon Intercomparison) studies (Scott et al. Reference Scott, Cook, Naysmith, Bryant and O’Donnell2007, Reference Scott, Cook and Naysmith2010a, Reference Scott, Cook and Naysmith2010b). Each AMS batch contains 13 humic acid samples, which are averaged to give a mean age for the batch. The standard deviation on these values is used to determine the error reported on the unknown samples in the batch.
An additional secondary standard generated from a single year’s growth of barley mash from the Glengoyne distillery. This was Sample A in the Third International Radiocarbon Intercomparison (TIRI). It is prepared by combusting a sufficient quantity of barley mash to generate 2 L of CO2, which is stored in a glass bulb. An aliquot of this gas is taken and converted to graphite to give an accurate check of the performance of the graphitization and AMS process (Naysmith et al. Reference Naysmith, Cook, Freeman, Scott, Anderson, Xu, Dunbar, Muir, Dougans, Wilcken, Schnabel, Russell, Ascough and Maden2010).
Significant quantities of bone are routinely measured within batches, and as a result, in addition to the mammoth background bone, a known-age in-house tertiary standard is also measured routinely to promote confidence in the isolation and analysis of the collagen fraction. This bone was supplied by English Heritage and has produced a mean laboratory age of 2132±42 14C yr BP since its first analysis in 2003 until 2012. A second bone, provided by Historic Scotland, has been used since 2012 and has produced a mean laboratory age of 427±40 14C yr BP.
The consensus values for both the secondary and tertiary standards used in the laboratory are shown in Tables 1 and 2. The consensus values from the intercomparison studies are also listed.
Table 1 Fraction modern values for different background types over the period from 2011 to 2014.

Table 2 Secondary standard 14C ages or F14C values over the period from 2011 to 2014.

AMS Analysis and Data Reduction
The SUERC operates two AMS instruments capable of making routine carbon measurements: a National Electrostatics Corporation (NEC) 5MV tandem accelerator mass spectrometer and a 250 kV single-stage accelerator mass spectrometer (SSAMS) both use 134-position MC-SNICS sources for running samples. Two sources are attached to the NEC 5MV Tandem AMS and one to the 250 kV single stage AMS. Batches of samples are notionally divided into 13 groups of 10 samples, with each group having 3 standards (one oxalic acid II primary standard, one humic acid secondary standard, and either a barley mash or a background secondary standard) and 7 unknowns.
As a means of monitoring the background of the AMS instrument, 1.5 mg of high-purity natural graphite powder, 100 mesh with a purity of 99.9995%, sourced from Alfa Aesar, are pressed into cathodes and routinely measured in the last two positions in every batch. These samples are of infinite age with respect to 14C. They have not undergone any in-house chemical processing and therefore do not contain any carbon contamination that may possibly be introduced during the chemical pretreatment processes. They thus represent the minimum 14C background value that can be obtained by the AMS instrument at the time. The measured 14C/13C ratios of graphite are generally less than 5×10–14 (equivalent to a 14C age of 65 kyr), which clearly shows that machine background and source cross-contamination are negligible.
All cathodes are repeatedly measured in intragroup rotation until both the counting statistics on each sample and the scatter on the 14C/13C ratio achieve a quality goal. Each primary standard is typically measured to a precision of 2.5‰, whereas the secondary standards (bulk TIRI barley mash and individual combusted humic acid) and unknown samples are routinely measured to a precision of 3‰. Measurement on such a group-by-group basis means that individual samples are less susceptible to long-term drifts in the machine tuning, while the inclusion of standards within each group allows for the monitoring and later correction of shorter-term changes. The individual combusted secondary standards are used to assess the total uncertainties on the measurements.
In the case where no temporal trend across the batch is apparent in the oxalic acid II primary standards and secondary standards, the NEC data analysis program abc is used to examine the measurements on each cathode, exclude aberrant measurements (for example, those made on the surface of the cathode during the initial “warm up” or those attempted after the expiration of the sample material within the cathode), then provide a weighted average ratio for each cathode.
Where a trend can be identified in the primary and secondary standards, an additional prenormalization step is applied in abc before producing a cathode average. Each individual measurement of the unknowns and secondary standards is normalized using the n temporally closest OxII measurements. The secondary standards are used as a guide to determine an appropriate number for n. In both cases, a final normalization using the average primary standard ratio is applied prior to age calculation.
Background Subtraction Calculation of 14C Ages
Prior to 2006, we calculated the final 14C results using the method set out in Donahue et al. (Reference Donahue, Linick and Jull1990). This method uses the raw 14C/13C ratios and then applies a fractionation factor calculated from the offline δ13C value. It then calculates fraction modern (Fm) values for the samples before applying a contamination correction to calculate the final F (F is Fm corrected for background) values that are used to calculate 14C ages. We used this method as it was consistent with the method used when we sent samples to the NSF-AMS facility in Arizona for measurement.
In 2007, SUERC altered the way in which 14C results are calculated (Brown and Southon Reference Brown and Southon1997). The background subtraction method uses the raw 14C/13C ratios and determines an average background value for the wheel from all the background samples. The next step is to subtract this ratio from all the standards and unknowns in the wheel. Fractionation correction is then applied to all unknowns and standards (using the fractionation factor determined from the offline δ13C) to calculate F.
Stable Isotope Measurements
13C analysis of subsamples of CO2 are measured using a VG SIRA 11 IRMS, comparing sample values with those of a working standard reference gas of known isotopic composition produced from international reference materials NBS19 and IAEA-CO-1. The measurement results are expressed using the δ notation (Craig Reference Craig1957) as per mil deviations from the VPDB standard, with 1σ precision of ±0.1‰. CO2 aliquots from the primary and secondary standards prepared daily are also measured. These values are used for offline normalization of sample 14C/13C ratios.
Continuous-Flow Isotope Ratio Mass Spectrometry (CF-IRMS)
δ13C, δ15N, and δ34S analyses are undertaken on bone collagen samples using a Thermo Scientific Delta V Advantage continuous-flow isotope ratio mass spectrometer (CF-IRMS) coupled via a Thermo Scientific Conflo IV to a Costech ECS 4010 elemental analyzer (EA) fitted with a pneumatic autosampler (Sayle et al. Reference Sayle, Cook, Ascough, Hastie, Einarsson, McGovern, Hicks, Edwald and Friðriksson2013).
In terms of δ13C and δ15N analyses, for every 10 unknown samples, in-house gelatin standards, which are calibrated to the international reference materials USGS40, USGS41, IAEA-CH-6, USGS25, IAEA-N-1, and IAEA-N-2, are run in duplicate. Results are again reported as per mil (‰) relative to the international standards VPDB and AIR with precisions of ±0.2‰ and ±0.3‰ for δ13C and δ15N, respectively. Results for samples with C/N ratios outside the range of 2.9–3.6 are discarded as they are deemed to represent collagen that had undergone postdepositional alteration (DeNiro and Hastorf Reference DeNiro and Hastorf1985). Supplementary analyses of our in-house mammoth bone background sample and our in-house standard bone are also routinely measured to check the consistency of the bone collagen separation chemistry. For δ34S analysis, two internal standards, which are calibrated to the international reference materials IAEA-S-1, IAEA-S-3, and IAEA-S-4, are run for every five unknown samples. Results are reported as per mil (‰) relative to the internationally accepted standard VCDT. The precision on the δ34S results is ±0.6‰. Approximately 25% of δ34S analyses are carried out in duplicate to verify the reproducibility.
ACKNOWLEDGMENTS
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors. The authors express their sincere thanks to Nicola Russell, Philippa Ascough, Graham Muir, Kerry Sayle, Helen Hastie, Terry Donnelly, Kerrie-Anne Lang, Iain Murdoch, and Linzi Straub.