Introduction
Tobacco-related illnesses have been estimated to claim 435 000 lives annually, thus constituting a serious and costly public health issue (Mokdad et al. Reference Mokdad, Marks, Stroup and Gerberding2004). Twin studies suggest a substantial genetic component to the development of nicotine dependence (e.g. Carmelli et al. Reference Carmelli, Swan, Robinette and Fabsitz1990; Kendler et al. Reference Kendler, Neale, Sullivan, Corey, Gardner and Prescott1999; True et al. Reference True, Xian, Scherrer, Madden, Bucholz, Heath, Eisen, Lyons, Goldberg and Tsuang1999), and a recent meta-analysis reported heritability estimates for smoking initiation ranging from 37 to 55% and persistent smoking ranging from 46 to 59% (Li et al. Reference Li, Cheng, Ma and Swan2003).
Given such evidence, several genome-wide linkage analyses have been conducted for a variety of tobacco-related phenotypes [e.g. DSM-IV nicotine dependence (APA, 1994), smoking quantity, and the Fagerström Nicotine Dependence Scale (Heatherton et al. Reference Heatherton, Kozlowski, Frecker and Fagerström1991)] to identify susceptibility loci related to nicotine dependence (Li, Reference Li2008). The earliest studies were conducted using an extended family design in the Collaborative Studies on the Genetics of Alcoholism (COGA) sample (Duggirala et al. Reference Duggirala, Almasy and Blangero1999) and a sib-pair design in samples collected in Christchurch, New Zealand and Richmond, VA, USA (Straub et al. Reference Straub, Sullivan, Ma, Myakishev, Harris-Kerr, Wormley, Kadambi, Sadek, Silverman, Webb, Neale, Bulik, Joyce and Kendler1999). Following these initial studies, more than 20 genome-wide linkage scans of tobacco-related phenotypes have been published, and several susceptibility regions have been identified across studies, including regions on chromosomes 9, 10, 11 and 17 (Li, Reference Li2008).
Association studies have begun to identify specific genes that underlie these linkage regions. For example, three independent studies have reported evidence of linkage between nicotine-related phenotypes and a region of chromosome 17 [20–65 centiMorgans (cM)] (Duggirala et al. Reference Duggirala, Almasy and Blangero1999; Wang et al. Reference Wang, Ma and Li2005; Li et al. Reference Li, Ma, Payne, Lou, Zhang, Dupont and Elston2008). A recent candidate gene study suggested polymorphisms in the nicotinic acetylcholine β1 receptor subtype gene (CHRNB1) may be responsible for this linkage signal (Lou et al. Reference Lou, Ma, Payne, Beuten, Crew and Li2006). Additionally, studies conducted using the COGA sample have reported evidence of linkage between nicotine-related phenotypes and chromosome 2 at 85 cM (Bergen et al. Reference Bergen, Korczak, Weissbecker and Goldstein1999; Bierut et al. Reference Bierut, Rice, Goate, Hinrichs, Saccone, Foroud, Edenberg, Cloninger, Begleiter, Conneally, Crowe, Hesselbrock, Li, Nurnberger, Porjesz, Schuckit and Reich2004). This region includes the neurexin 1 gene (NRXN1), which contains polymorphisms associated with nicotine dependence in a recent genome-wide association study (GWAS) (Bierut et al. Reference Bierut, Madden, Breslau, Johnson, Hatsukami, Pomerleau, Swan, Rutter, Bertelsen, Fox, Fugman, Goate, Hinrichs, Konvicka, Martin, Montgomery, Saccone, Saccone, Wang, Chase, Rice and Ballinger2007) as well as recent candidate gene studies (Nussbaum et al. Reference Nussbaum, Xu, Payne, Ma, Huang, Gelernter and Li2008; Sherva et al. Reference Sherva, Wilhelmsen, Pomerleau, Chasse, Rice, Snedecor, Bierut, Neuman and Pomerleau2008). These studies demonstrate the progress being made in understanding the genetic contributions to tobacco-related phenotypes and how linkage analysis has aided in the identification of susceptibility loci for such traits.
In addition to identifying susceptibility loci for nicotine dependence specifically, several studies have tried to identify loci that contribute to substance dependence more generally by searching for genomic regions that confer risk to both nicotine and alcohol dependence. Such studies are justified by twin studies indicating a shared genetic etiology between these two disorders (Swan et al. Reference Swan, Carmelli and Cardon1997; True et al. Reference True, Xian, Scherrer, Madden, Bucholz, Heath, Eisen, Lyons, Goldberg and Tsuang1999), and several have reported positive findings. For example, data from the COGA sample suggest that the susceptibility locus on chromosome 2 containing NRXN1 contributed to both nicotine and alcohol dependence (Bergen et al. Reference Bergen, Korczak, Weissbecker and Goldstein1999). Further, a study of the Mission Indian population found that the chromosome 4 region containing the alcohol dehydrogenase gene cluster contributed to increased risk for both disorders (Ehlers & Wilhelmsen, Reference Ehlers and Wilhelmsen2006). In addition, a Finnish twin sample was used to identify loci on chromosomes 7 and 11 (Loukola et al. Reference Loukola, Broms, Maunu, Widen, Heikkila, Siivola, Salo, Pergadia, Nyman, Sammalisto, Perola, Agrawal, Heath, Martin, Madden, Peltonen and Kaprio2008) and sibling pairs collected in Ireland were used to identify loci on chromosomes 7 and 18 (Sullivan et al. Reference Sullivan, Kuo, Webb, Neale, Vittum, Furberg, Walsh, Patterson, Riley, Prescott and Kendler2008) conferring risk for both disorders. Such studies provide important insights into how different chromosomal regions confer risk to substance dependence whether it is towards a specific substance or towards a more general tendency toward addictive behavior.
The current study conducted a genome-wide linkage scan for nicotine dependence in the University of California, San Francisco (UCSF) Family Alcoholism Study to support and extend previous findings. Linkage peaks were followed-up by analysing each of the 14 nicotine dependence symptoms assessed by the Semi-Structured Assessment for the Genetics of Alcoholism (SSAGA; Bucholz et al. Reference Bucholz, Cadoret, Cloninger, Dinwiddie, Hesselbrock, Nurnberger, Reich, Schmidt and Schuckit1994) to identify those symptoms responsible for the reported linkage signals. A further aim was to determine whether the linked genomic regions contributed to nicotine dependence specifically, or whether they might confer increased risk to addiction more generally by showing evidence of linkage to both alcohol and nicotine dependence. Thus, supplementary genome-wide linkage scans of nicotine dependence were conducted utilizing alcohol dependence diagnoses alternatively as a covariate and as an additional predictor in a bivariate analysis.
Method
Participants
The present study utilized participants from the UCSF Family Alcoholism Study (Seaton et al. Reference Seaton, Cornell, Wilhelmsen and Vieten2004; Vieten et al. Reference Vieten, Seaton, Feiler and Wilhelmsen2004), which consists of 2524 participants from 890 families (average size=2.83 members). The UCSF study was a nationwide study on the genetics of alcoholism and other substance dependence designed to recruit a large number of small family pedigrees enriched for alcohol dependence. Probands were sampled from the community and invited to participate if they met screening criteria for alcohol dependence at some point in their lifetime and had at least one sibling or both parents available to participate. Probands were excluded if they reported serious drug addictions (defined as use of stimulants, cocaine, or opiates daily for >3 months or weekly for >6 months), any history of intravenous substance use, a current or past diagnosis of schizophrenia, bipolar disorder, or other psychiatric illness involving psychotic symptoms (those with depressive and anxiety disorders were accepted), a life-threatening illness, or an inability to speak and read English. Relatives of qualifying probands were invited by mail to participate.
The UCSF Family Alcoholism Study sample consisted of 1548 women and 976 men with a mean age of 48.5 (s.d.=13.4) years. The mean educational level of the sample was 14.3 (s.d.=2.9) years, and the mean annual income was US$ 54672 (s.d.=53421) (median, US$ 45000). The racial distribution was 92% Caucasian, 3% each African-American and Hispanic, and 1% each Native American and other. No attempt was made to exclude or over-sample minorities. A total of 365 participants (15%) were diagnosed with nicotine dependence only, 464 (18%) were diagnosed with alcohol dependence only, and 880 (35%) were diagnosed with both disorders.
An unselected general population sample of 147 individuals was recruited to assess phenotype base rates. Letters were sent to residents of the same geographical areas as the family samples, requesting participation in a study on ‘health behaviors and characteristics’ to avoid a sample biased toward participation in a study on alcoholism. No inclusion or exclusion criteria were applied aside from the ability to respond to the telephone interview and complete the questionnaires. Within this population, 36 participants (24%) were diagnosed only with nicotine dependence, 14 (10%) were diagnosed only with alcohol dependence, and 11 (7%) were diagnosed with both disorders. The recruitment details of all participants have been previously published (Seaton et al. Reference Seaton, Cornell, Wilhelmsen and Vieten2004; Vieten et al. Reference Vieten, Seaton, Feiler and Wilhelmsen2004).
All participants were administered a modified version of the SSAGA (Bucholz et al. Reference Bucholz, Cadoret, Cloninger, Dinwiddie, Hesselbrock, Nurnberger, Reich, Schmidt and Schuckit1994), an interview developed by COGA, which was used to diagnose DSM-IV alcohol and other substance dependence and abuse and to collect demographic, medical, psychiatric, alcohol, nicotine, and other drug-use history. This modified version of the SSAGA included questions assessing each of the DSM-IV substance dependence symptoms as applied to tobacco use as well as a question assessing whether these symptoms occurred within a 12-month period to allow for the assignment of DSM-IV diagnoses of nicotine dependence. Participants that did not meet criteria for DSM-IV nicotine dependence included those that had never smoked as well as those that had smoked but did not meet full diagnostic criteria. Only sections of the SSAGA assessing DSM-IV alcohol and substance misuse diagnoses were administered due to time constraints.
Deoxyribonucleic acid (DNA) collection and genotyping
The DNA extraction procedure and genotyping protocol have been previously described (Wilhelmsen et al. Reference Wilhelmsen, Schuckit, Smith, Lee, Segall, Feiler and Kalmijn2003). Briefly, DNA was isolated from whole blood using the Puregene kit (Gentra Systems, USA), and genotypes for a panel of microsatellite polymorphisms were generated using fluorescently labeled polymerase chain reaction primers (HD5, version 2.0; Applied Biosystems, USA). The HD5 panel set consisted of 811 markers with an average marker-to-marker distance of 4.6 cM (maximum, 14 cM) and an average heterozygosity of greater than 77%. The sizes of marker amplimers were determined (blinded to pedigree structure and subject characteristics) from the electropherogram using the Genotyper software package (Applied Biosystems, USA). All electropherograms were visually inspected and exported from Genotyper in base pair sizes relative to the standard measured to one hundredth of a base pair. Allele frequencies observed in the founders were used for all analyses. The sex-averaged marker map order obtained from the manufacturer was used and verified with the family data from the current sample.
Prior to analysis, a number of quality-control steps were undertaken to ensure the accuracy of the genotype calls. First, the pedigree relationship statistical test (McPeek & Sun, Reference McPeek and Sun2000) software was used to identify sample and pedigree structure errors based on the genotypes for autosomal markers. For those individuals in which a probable error was detected, DNA was re-isolated from a stored frozen blood specimen and the genotyping was repeated. If the error could not be resolved, the problematic genotype was subsequently treated as missing. A total of 15 families were identified with pedigree structure errors. Of these, five were resolved following re-genotyping. Second, Mendelian errors were identified using the program pedcheck (O'Connell & Weeks, Reference O'Connell and Weeks1998). When isolated Mendelian errors were observed, the genotypes for the entire family were excluded for the marker yielding the error. Markers exhibiting a high rate of Mendelian errors across families were excluded from subsequent analysis. pedcheck identified 3104 Mendelian errors resulting in 7714 lost genotypes and the exclusion of one marker. Third, the error-checking algorithm implemented in Merlin (Abecasis et al. Reference Abecasis, Cherny, Cookson and Cardon2002) was used to assess the probability that each genotype was correctly called. A total of 1867 genotypes with probabilities of less than 0.025 of being correct were removed from further analysis. Following these quality-control procedures, a total of 1 269 708 genotypes were accepted for analysis with a success rate of 99.6%.
Analysis
Both genotype and phenotype data were available for 1647 individuals, and phenotype but not genotype data were available for 875 individuals. Participants not genotyped included relatives of genotyped probands that did not provide DNA samples and probands and their relatives not belonging to a genetically informative pedigree. Notably, rates of alcohol and nicotine dependence remained unchanged when participants not genotyped were removed from the sample (data not shown). Seven hundred and thirteen families that contained sibling, half-sibling, avuncular or cousin pairs were considered genetically informative for linkage analysis. These families ranged in size from three to 20 subjects [average 4.63 (s.d.=2.13)]. The data include: 1085 sibling, 40 half-sibling, 17 grandparent–grandchild, 238 avuncular and 32 cousin relative pairs. An additional 177 families contained only a single individual with phenotype data. These individuals were included within some analyses to the extent that they contribute information about trait means and variance and the impact of covariates.
Initial analyses were conducted to determine the potential of the DSM-IV nicotine dependence diagnosis and each of its 14 dichotomously scored symptoms as assessed by the SSAGA to exhibit evidence of linkage. Using a variance components approach, SOLAR (Almasy & Blangero, Reference Almasy and Blangero1998) estimates h2 by partitioning the trait relative-pair covariance into additive genetic and environmental contributions while correcting for participants' age and sex. The probability that h2 was greater than zero was determined using a Student's t test, which was used to evaluate the potential of the nicotine dependence diagnosis and its constituent symptoms to detect linkage.
The variance components approach to linkage incorporates information from affected and unaffected participants by estimating linkage across the entire pedigree (Almasy & Blangero, Reference Almasy and Blangero1998), and has been shown to possess comparable statistical power compared with relative-pair-based methods [i.e. Kong & Cox (Reference Kong and Cox1997) statistic] for both quantitative (Amos et al. Reference Amos, Krushkal, Thiel, Young, Zhu, Boerwinkle and de Andrade1997) and dichotomous phenotypes (Duggirala et al. Reference Duggirala, Williams, Williams-Blangero and Blangero1997). Thus, the variance components approach implemented in SOLAR was used to calculate multipoint logarithm (base 10) of odds (LOD) scores across the genome at 1-cM intervals for DSM-IV nicotine dependence. Peaks that achieved a LOD score >1.0 were reported as regions of interest as suggested by Lander & Kruglyak (Reference Lander and Kruglyak1995).
Dichotomous phenotypes were modeled as latent normally distributed variables with a threshold above which an individual was considered ‘affected’. It is notable that when applied to dichotomous phenotypes, variance components linkage analysis can yield inflated LOD scores if distributional assumptions are violated (Duggirala et al. Reference Duggirala, Williams, Williams-Blangero and Blangero1997). To protect against this potential bias, allele-sharing probabilities for a simulated locus under the null hypothesis of no linkage were generated across 250 000 trials (Duggirala et al. Reference Duggirala, Blangero, Almasy, Arya, Dyer, Williams, Leach, O'Connell and Stern2001), an approach shown to yield appropriate type I error rates (Jung et al. Reference Jung, Weeks and Feingold2006). The distribution of simulated LOD scores was used to calculate point-wise estimates of significance according to the formula p=(r+1)/(n+1), where r=the number of simulated values greater than the observed value and n=the number of simulations (North et al. Reference North, Curtis and Sham2002). As a further protection, relative-pair analyses utilizing the Kong & Cox (Reference Kong and Cox1997) statistic as implemented in Merlin (Abecasis et al. Reference Abecasis, Cherny, Cookson and Cardon2002) were conducted for each reported linkage peak to test whether evidence for linkage was robust across analytic methods. All p values were interpreted using guidelines suggested by Lander & Kruglyak (Reference Lander and Kruglyak1995).
SOLAR also allows for the simultaneous analysis of multiple traits as well as the inclusion of multiple covariates. In the bivariate case, SOLAR estimates the proportion of variance in each phenotype that can be explained by a genetic locus as well as estimating the proportion of the genetic correlation between the phenotypes that can be explained by this locus. To ensure that reported linkage peaks were not the result of a small subset of pedigrees, homogeneity tests were performed using the SOLAR hlod function (Goring, Reference Goring2002). This test contrasts a null model in which families belong to a single distribution exhibiting genetic linkage to the tested locus against an alternative model in which families belong to one of two distributions, only one of which shows evidence of genetic linkage to the tested locus.
For the linkage analysis of the 14 SSAGA nicotine dependence symptoms, the support intervals for reported linkage peaks were defined as the region surrounding a linkage peak yielding a LOD score that was greater than the maximum LOD – 1 in each direction. Only these support intervals were subjected to linkage analysis to reduce the number of statistical tests. Further, LOD scores were only interpreted as a measure of magnitude and were not used to determine significance given the exploratory and descriptive nature of these analyses. Because the current sample was selected for alcohol dependence, which is highly correlated with tobacco use (Miller & Gold, Reference Miller and Gold1998), prevalence rates for nicotine dependence and its respective symptoms were estimated in the unselected control sample and included in the tested models to correct for ascertainment bias when calculating h2 and linkage. Finally, allele frequencies used to calculate allele-sharing probabilities between relative pairs were estimated using study participants from varying ethnic backgrounds. This approach can lead to biased allele frequency and sharing probabilities amongst minority participants. Thus, analyses were conducted for the full sample as well as restricting the sample to only Caucasian participants. To eliminate the potential for the described bias, results for the Caucasian-only sample are reported, though notably only small changes were observed in LOD scores across analyses (ΔLOD<0.05 for the variance components analysis and ΔLOD<0.4 for the relative-pair analysis).
Results
The h2 estimate for the nicotine dependence diagnosis was significantly greater than 0 (p<0.001), as were the h2 estimates for 13 of the 14 nicotine dependence symptoms (p<0.05). The remaining symptom, ‘continued smoking despite physical illness’, approached significance (p=0.08).
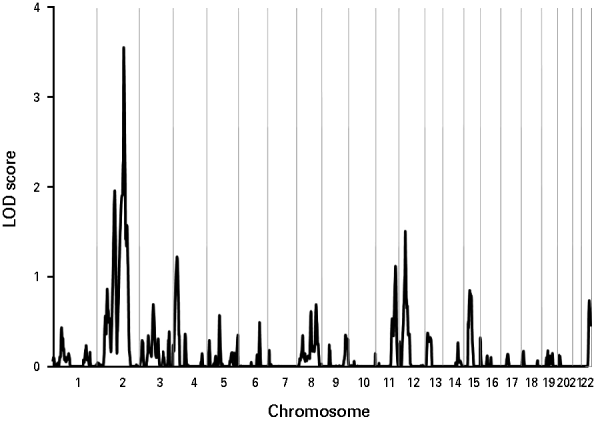
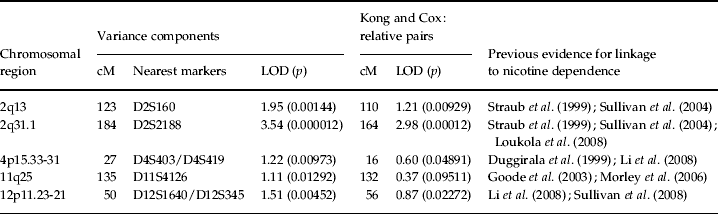
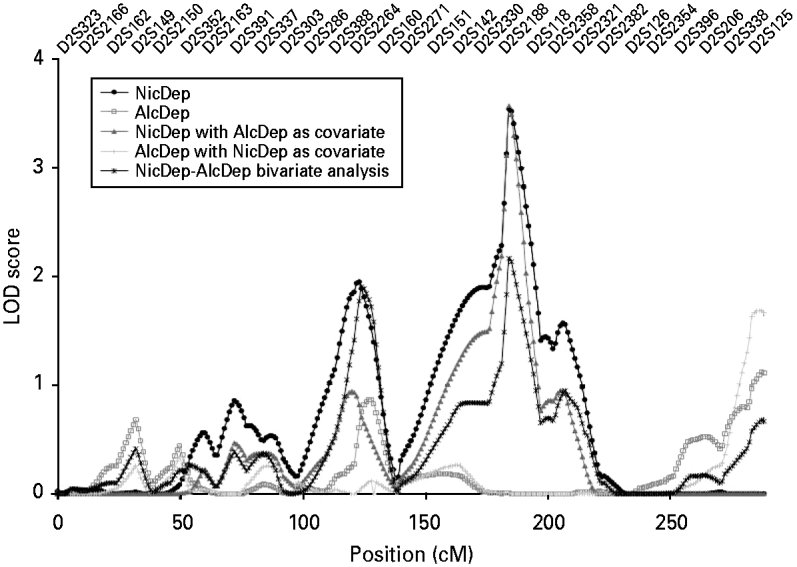
Variance components linkage analysis of the nicotine dependence diagnosis yielded evidence for linkage on chromosome 2 at 184 cM nearest to marker D2S2188 with an associated LOD score of 3.54 (Fig. 1), which achieved genome-wide significance (point-wise empirical p=0.000012) (see Table 1). The parallel relative-pair analysis also revealed a linkage peak in this region at 164 cM nearest to marker D2S151 that failed to reach genome-wide significance (all pairs: LOD=2.98, point-wise empirical p=0.00012), but, nonetheless, suggested the evidence for linkage to this region was robust across analytic methods. The shift in location of the linkage peaks likely resulted from differences in LOD score calculations across methods given that the same identity-by-descent (IBD) estimates were used for both sets of analyses.

Fig. 1. Multipoint linkage analysis for nicotine dependence for the entire genome. Chromosome numbers are represented on the x axis, and logarithm (base 10) of odds (LOD) scores are represented on the y axis. Results for each chromosome are aligned end to end with the p-terminus on the left. Vertical lines indicate the boundaries between chromosomes.
Table 1. Chromosomal regions with evidence of linkage to DSM-IV nicotine dependence
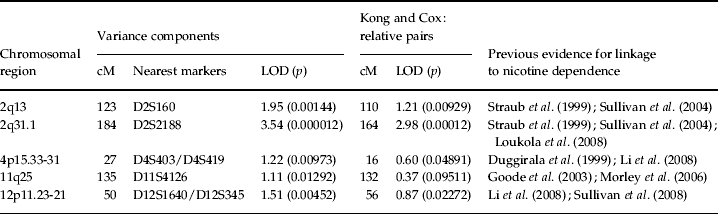
cM, centiMorgans; LOD, logarithm (base 10) of odds.
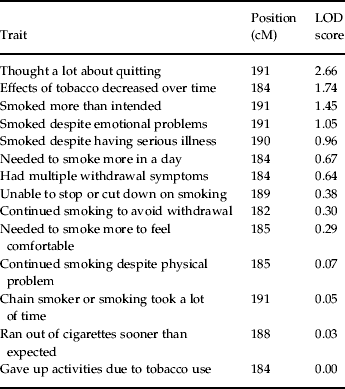
Notably, there was no evidence of heterogeneity in LOD scores across pedigrees, as each family had an estimated α, interpreted as the probability of a given family belonging to a single population yielding evidence for linkage, that was >0.99 for each peak as estimated by the SOLAR hlod function (Goring, Reference Goring2002). Linkage analysis of the 14 nicotine dependence symptoms assessed by the SSAGA across the support interval of this peak (181–192 cM) showed that the symptom ‘thought a lot about quitting or cutting down’ yielded the strongest evidence for linkage (LOD=2.66). The symptoms ‘tobacco had less effect with continued regular use’ (LOD=1.74), ‘found self smoking more than intended’ (LOD=1.45), and ‘continued to smoke despite tobacco-induced emotional problems’ (LOD=1.05) also appeared to contribute to the observed linkage peak (for complete results, see Table 2).
Table 2. LOD scores for symptoms at linkage peak on chromosome 2 at 184 cM
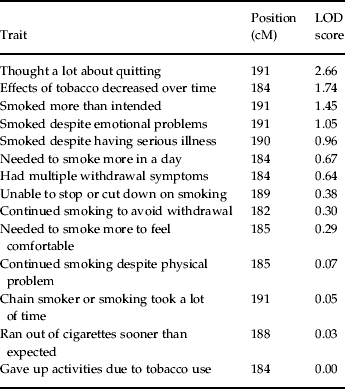
LOD, Logarithm (base 10) of odds; cM, centiMorgans.
Four additional loci yielded notable evidence of linkage (Table 1). The first locus represented a second peak located on chromosome 2 at 123 cM nearest to marker D2S160 (LOD=1.95, point-wise empirical p=0.00144) with a support interval that extended from 109 to 132 cM. The second locus was found on chromosome 4 at 27 cM nearest to markers D4S403 and D4S419 (LOD=1.22, point-wise empirical p=0.00973) with a support interval that extended from 7 to 43 cM. The third peak was found on chromosome 11 at 135 cM nearest to marker D11S4126 (LOD=1.11, point-wise empirical p=0.01292) with a support interval that spanned from 106 to 146 cM, and the fourth locus was found on chromosome 12 at 50 cM nearest to markers D12S1640 and D12S345 (LOD=1.51, point-wise empirical p=0.00452) with a support interval that extended from 37 to 67 cM. Among these peaks, the parallel relative-pair analyses only found evidence of linkage to the peak on chromosome 2 at 123 cM, though this evidence was fairly weak (LOD=1.21, point-wise empirical p=0.00929).
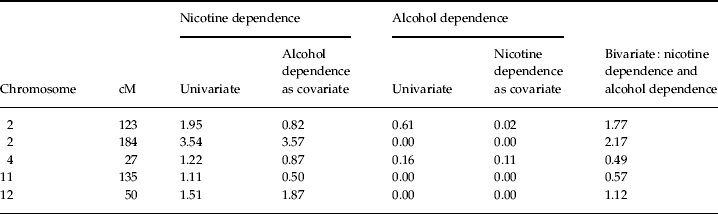
Linkage results for the nicotine dependence diagnosis were then compared with the results from a previous linkage scan of alcohol dependence. None of the regions reported in the previous scan yielded evidence of linkage to nicotine dependence. Additionally, when nicotine dependence was included as a covariate in a linkage analysis of alcohol dependence, LOD scores for the alcohol dependence diagnosis approached 0 in those regions that showed linkage to nicotine dependence in the present report (Table 3). These results provide evidence suggesting that the regions reported here confer risk towards nicotine but not alcohol dependence.
Table 3. LOD scores for linkage analysis of nicotine and alcohol dependence phenotypes
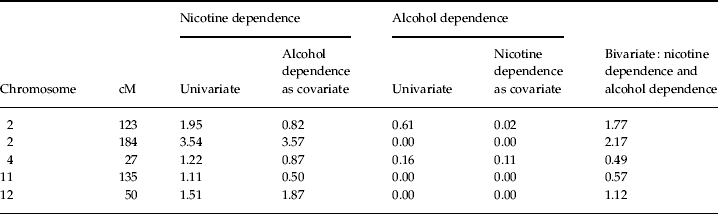
LOD, Logarithm (base 10) of odds; cM, centiMorgans.
This conclusion was supported by subsequent analyses in which the alcohol dependence diagnosis was included in the linkage analysis of nicotine dependence first as a covariate and then as a second phenotype in a bivariate analysis. For the reported peak on chromosome 2 at 184 cM, a slight increase in the initial LOD score was observed when the alcohol dependence diagnosis was added as a covariate (from 3.54 to 3.57), whereas a drop in the LOD score was observed for the bivariate analysis (from 3.54 to 2.17) that probably occurred because of corrections for the inclusion of additional parameters. This result suggests that this region confers risk towards nicotine but not alcohol dependence (see Fig. 2). Similar conclusions were made regarding the remaining peaks of interest (for complete results, see Table 3).
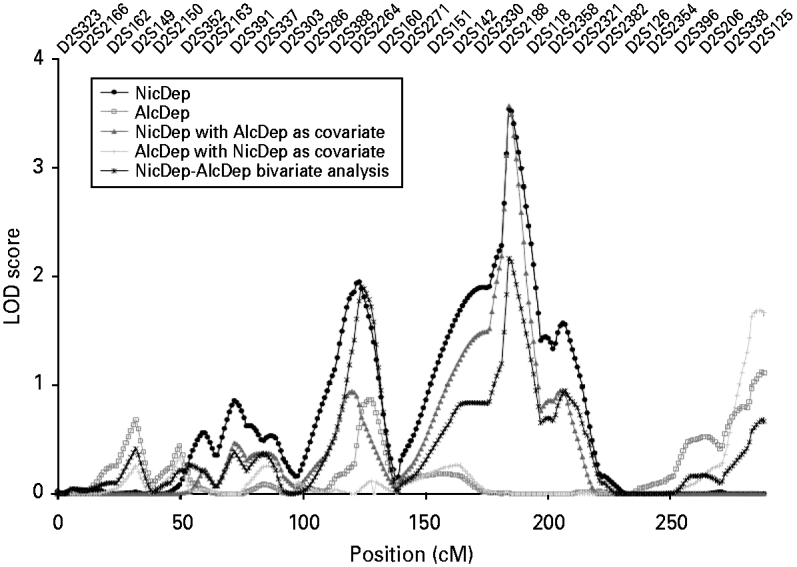
Fig. 2. Linkage analysis for the nicotine and alcohol dependence phenotypes on chromosome 2. LOD, logarithm (base 10) of odds; cM, centiMorgans; NicDep, nicotine dependence; AlcDep, alcohol dependence.
Discussion
The primary aim of the current study was to conduct a genome-wide linkage scan of nicotine dependence in the UCSF Family Alcoholism Study sample to support and extend the findings of previous studies. A linkage peak was observed on chromosome 2 at 184 cM that achieved genome-wide significance when a variance components approach was used based on criteria described by Lander & Kruglyak (Reference Lander and Kruglyak1995) . This region continued to yield strong evidence for linkage when the Kong & Cox (Reference Kong and Cox1997) statistic was used to test for linkage, but failed to reach genome-wide significance. This divergence in the results limits claims of genome-wide significance, though the consistency in LOD scores across analytic methods provides strong evidence of linkage to this region. Additional peaks of interest were found on a second region of chromosome 2 at 123 cM, chromosome 4 at 27 cM, chromosome 11 at 135 cM, and chromosome 12 at 50 cM, though these should be interpreted tentatively given the weaker evidence for linkage.
The linkage region on chromosome 2 at 184 cM has been previously identified as harboring a susceptibility locus for nicotine dependence (Straub et al. Reference Straub, Sullivan, Ma, Myakishev, Harris-Kerr, Wormley, Kadambi, Sadek, Silverman, Webb, Neale, Bulik, Joyce and Kendler1999; Loukola et al. Reference Loukola, Broms, Maunu, Widen, Heikkila, Siivola, Salo, Pergadia, Nyman, Sammalisto, Perola, Agrawal, Heath, Martin, Madden, Peltonen and Kaprio2008). The first study reported a linkage signal approximately 35 megabases centromeric, and the second study reported a linkage signal approximately 32 megabases telomeric of the peak reported in this study. In addition, a locus on the short arm of chromosome 2 at 85 cM, which contains NRXN1, has been previously linked to both nicotine and alcohol dependence (Yang et al. Reference Yang, Chang, Lin, Chen, Lin and Fann2005; Bierut et al. Reference Bierut, Madden, Breslau, Johnson, Hatsukami, Pomerleau, Swan, Rutter, Bertelsen, Fox, Fugman, Goate, Hinrichs, Konvicka, Martin, Montgomery, Saccone, Saccone, Wang, Chase, Rice and Ballinger2007; Nussbaum et al. Reference Nussbaum, Xu, Payne, Ma, Huang, Gelernter and Li2008), but this locus is approximately 100 megabases from the locus identified in the present study.
There are potential candidate genes within the support interval of the locus reported here. For example, the nicotinic acetylcholine α1 gene (CHRNA1) is located near the center of the reported linkage peak. Although originally thought to be found only in muscle tissue, recent gene expression studies have found this gene to be expressed in brain as well (Su et al. Reference Su, Wiltshire, Batalov, Lapp, Ching, Block, Zhang, Soden, Hayakawa, Kreiman, Cooke, Walker and Hogenesch2004), suggesting a potential role in nicotine addiction. Evidence for an association between this gene and smoking behavior has been previously reported (Faraone et al. Reference Faraone, Su, Taylor, Wilcox, Van Eerdewegh and Tsuang2004), but negative findings have also been described (Sherva et al. Reference Sherva, Wilhelmsen, Pomerleau, Chasse, Rice, Snedecor, Bierut, Neuman and Pomerleau2008). In addition, single nucleotide polymorphisms (SNPs) located in the nearby growth factor receptor-bound protein 14 (GRB14) and grancalcin (GCA) genes were recently associated with nicotine dependence in a GWAS (Vink et al. Reference Vink, Smit, de Geus, Sullivan, Willemsen, Hottenga, Smit, Hoogendijk, Zitman, Peltonen, Kaprio, Pedersen, Magnusson, Spector, Kyvik, Morley, Heath, Martin, Westendorp, Slagboom, Tiemeier, Hofman, Uitterlinden, Aulchenko, Amin, van Duijn, Penninx and Boomsma2009). Potential mechanisms through which one or both of these genes might confer risk to nicotine dependence are not clear. GRB14 is thought to be involved in insulin receptor signaling and may influence signaling pathways that regulate growth and metabolism (Carre et al. Reference Carre, Cauzac, Girard and Burnol2008). GCA may be involved in the migration and adherence of neutrophils (Jia et al. Reference Jia, Han, Borregaard, Lollike and Cygler2000). Thus, further studies are necessary to determine whether a causal variant is located in GRB14 or GCA or whether the associated SNPs are in linkage disequilibrium with a causal variant located in a nearby gene such as CHRNA1.
Follow-up analyses of the chromosome 2 linkage peak showed that SSAGA symptoms encompassing a broad range of DSM-IV nicotine dependence symptom clusters, including evidence of tolerance, inability to quit smoking, escalating pattern of use, and persistent use despite negative health consequences, provided modest contributions to the linkage signal. This suggests that the chromosome 2 locus at 184 cM confers risk for nicotine dependence in general rather than a specific facet of this disorder. As further evidence of this conclusion, it is notable that the linkage analysis of the nicotine dependence diagnosis yielded a higher LOD score than any of the individual nicotine dependence symptoms (LOD=3.54 v. maximum LOD=2.66). These results are consistent with a previous study demonstrating that a single genetic factor can explain a predominant proportion of the common variation between DSM-IV nicotine dependence symptoms (Lessov et al. Reference Lessov, Martin, Statham, Todorov, Slutske, Bucholz, Heath and Madden2004).
A further aim of this study was to determine whether the reported genomic regions contributed to nicotine dependence specifically or to addiction more generally by showing evidence of linkage to both alcohol and nicotine dependence. The former conclusion was supported, as no overlaps between linkage signals reported in the present study were observed with those reported in a previous linkage study of alcohol dependence to chromosomes 1 at 11 cM, 2 at 287 cM, 8 at 163 cM, and 18 at 48 cM (I. R. Gizer et al. unpublished observations). Further, supplementary genome-wide linkage scans of nicotine dependence utilizing alcohol dependence diagnoses alternatively as a covariate and as an additional predictor in a bivariate analysis showed little evidence of linkage between alcohol dependence and the regions reported herein. This suggests that the susceptibility loci identified in the present study are specifically involved in the etiology of nicotine dependence and are unrelated to alcohol dependence.
This result was somewhat surprising given the strong correlations between drinking and smoking behaviors (Miller & Gold, Reference Miller and Gold1998). Twin studies suggest that common genetic influences are partially responsible for the observed correlation (Swan et al. Reference Swan, Carmelli and Cardon1997; True et al. Reference True, Xian, Scherrer, Madden, Bucholz, Heath, Eisen, Lyons, Goldberg and Tsuang1999), though disorder-specific genetic influences have been identified as well (Kendler et al. Reference Kendler, Myers and Prescott2007; Volk et al. Reference Volk, Scherrer, Bucholz, Todorov, Heath, Jacob and True2007). Nonetheless, previous family-based samples selected for alcohol dependence have identified genetic loci that confer risk to alcohol- and tobacco-use phenotypes. For example, loci on chromosomes 2 (Bierut et al. Reference Bierut, Rice, Goate, Hinrichs, Saccone, Foroud, Edenberg, Cloninger, Begleiter, Conneally, Crowe, Hesselbrock, Li, Nurnberger, Porjesz, Schuckit and Reich2004), 4 (Ehlers & Wilhelmsen, Reference Ehlers and Wilhelmsen2006), 7 (Loukola et al. Reference Loukola, Broms, Maunu, Widen, Heikkila, Siivola, Salo, Pergadia, Nyman, Sammalisto, Perola, Agrawal, Heath, Martin, Madden, Peltonen and Kaprio2008; Sullivan et al. Reference Sullivan, Kuo, Webb, Neale, Vittum, Furberg, Walsh, Patterson, Riley, Prescott and Kendler2008) and 18 (Sullivan et al. Reference Sullivan, Kuo, Webb, Neale, Vittum, Furberg, Walsh, Patterson, Riley, Prescott and Kendler2008) have been shown to contribute jointly to alcohol and nicotine dependence. The lack of such findings in the present study suggests that unique genetic influences contributed to nicotine and alcohol dependence in the UCSF Family Alcoholism Study.
The reported findings have important implications for molecular genetic studies of nicotine dependence, but there are limitations that should be noted. For example, the UCSF Family sample was originally selected for alcohol dependence. Thus, it is not clear how the reported findings will generalize to populations without this bias, though a previous study reporting evidence of linkage to the chromosome 2 region used a sample selected for nicotine rather than alcohol dependence (Loukola et al. Reference Loukola, Broms, Maunu, Widen, Heikkila, Siivola, Salo, Pergadia, Nyman, Sammalisto, Perola, Agrawal, Heath, Martin, Madden, Peltonen and Kaprio2008). Additionally, categorizing ‘never smokers’ and ‘not nicotine-dependent’ participants as unaffected individuals may have influenced study results (Munafò et al. Reference Munafö, Clark, Johnstone, Murphy and Walton2004). Given that unique genetic influences contribute to the initiation and persistent use of tobacco (Heath & Madden, Reference Heath, Madden, Turner, Cardon and Hewitt1995), combining these participants into a single unaffected category likely limited our ability to detect these unique genetic influences. Nonetheless, there is substantial overlap in the genetic influences contributing to these stages of tobacco use (Sullivan & Kendler, Reference Kendler, Neale, Sullivan, Corey, Gardner and Prescott1999), providing justification for this approach. The statistical power of the present study is another possible limitation. Linkage studies lack sufficient statistical power for identifying loci with small effects, and this may explain the lack of support for loci such as chromosomes 9q and 10q that have been previously linked to nicotine dependence.
In summary, the current study adds to the literature by supporting evidence of genetic linkage of chromosome 2q to nicotine dependence. This study extends this finding by showing that this region confers risk to the full nicotine dependence diagnosis rather than a specific facet of the disorder. Finally, the present study suggests that this locus, as well as the additional loci identified, confers risk to nicotine but not alcohol dependence, thus providing evidence that this genomic region may harbor a gene specifically involved in the physiological effects and/or metabolism of nicotine.
Acknowledgements
The present study was supported by funds from the State of California for medical research on alcohol and substance abuse through the UCSF. Additional support was provided by the Ernest Gallo Clinic and Research Center (to K.C.W.), AA10201 and U54 RR0250204 (to C.L.E.) and T32 AA007573 (to I.R.G.).
Declaration of Interest
None.