


I. INTRODUCTION
Line profile analysis (LPA) is a typical application of diffraction providing microstructure information from a wide class of crystalline materials, ranging for example, from organic/pharmaceutical to ceramics and metals. Simple methods, some of them are known for almost a century, are described in traditional textbooks (Klug and Alexander, Reference Klug and Alexander1974; Schwartz and Cohen, Reference Schwartz and Cohen1977; Guinebretiere, Reference Guinebretiere2007; Dinnebier and Billinge, Reference Dinnebier and Billinge2008), and are routinely used by several laboratories of an order of magnitude estimate for example, of the domain size in nanocrystalline powders. The research is still strong in the field and important milestones have been reached in the past 20 years (Snyder et al., Reference Snyder, Fiala and Bunge2000; Mittemeijer and Scardi, Reference Mittemeijer and Scardi2004); full pattern methods have been developed considerably, to provide information that can nowadays easily complement electron microscopy observations.
It is often considered that LPA does not require highly sophisticated or advanced instruments for data collection, as the interest is in broadened line profiles. This is partly true, as it is relatively easy and straightforward to collect powder diffraction data and assess the peak profile width, but a proper evaluation of the data is still important, especially when accurate quantitative information is sought. For example, the instrumental profile function (IPF, the instrumental contribution to the observed diffraction line profile) should be carefully determined, especially when the width of the observed line profiles approaches the instrumental resolution. More significantly with synchrotron radiation (SR), beamlines can collect high-energy X-ray diffraction (XRD) data so as to encompass a large number of peaks and significantly increase the resolution and information content in a powder pattern; better statistics can also be relevant to appreciate fine details in the peak shape, and properly solve line profile overlapping.
With so many issues and given the variety of available LPA methods with the absence of a microstructure standard, the availability of a fully characterized reference material can be useful to test instrument features and performance, data collection, and reduction procedures.
Among the many possible reference candidates, a ball milled iron-alloy powder seems the ideal one for the possibility of assessing both domain size and strain contributions to the line profiles in the same powder pattern. The simple body centered cubic (bcc) crystal structure gives in fact relatively few peaks: this is of key importance to minimize peak overlapping and to provide, at the same time, intense, and easily measurable diffraction signals.
In the search for appropriate materials and operating conditions to produce a sufficiently large batch of ball milled powder, stability in time is a primary need and a basic starting point to develop a reference material. In the present work, the stability of the suggested material is demonstrated over a time range of 10 years, also providing a preliminary assessment of size/strain values that can be measured in a ball milled iron alloy. Standardization and scaling up to produce suitably large amounts of powder are then a relatively simple step forward.
II. EXPERIMENTAL
The metal powder of this study is a commercial Fe–1.5 wt% Mo alloy (Astaloy Mo®), easily available in large quantities and at a low cost, requiring no special care for handling and storage. Grinding was made in a planetary ball mill (Fritsch Pulverisette P4), using steel jars and 100Cr6 steel balls for 128 h. All operating parameters were suitably optimized in a previous study (D'Incau et al., Reference D'Incau, Leoni and Scardi2007), aiming at producing a uniform material with limited contamination from the grinding media. A lubricant (96% vol. ethanol) was added to prevent cold welding or sticking of the powder to the jar walls. To avoid oxidation, grinding was performed in a controlled Ar atmosphere (2% vol. O2max).
Powder diffraction laboratory data were collected on a Rigaku PMG-VH instrument, based on the traditional Bragg–Brentano configuration with a vertical flat-plate sample holder. A graphite bent crystal-analyzer in the diffracted beam was used to filter the Cu radiation produced by a sealed X-ray tube. Together with narrow slits (1° divergence slit, 2° soller slits, and 0.15 mm receiving slit), the optical setup of the diffractometer was such to produce narrow and symmetrical instrumental line profiles across the whole range of required diffraction angles. The IPF was evaluated experimentally with a LaB6 standard (Staudemann et al., Reference Staudemann2000), using pseudoVoigt (pV) functions to fit the instrumental line profiles. The trend of the full-width half-maximums (FWHM) was parameterized refining the coefficients of the well-known Caglioti's expression (Caglioti et al., Reference Caglioti, Paoletti and Ricci1958), whereas a simple polynomial was used to fit the trend of the Lorentzian fraction (mixing) parameter.
SR XRD data were collected at ESRF (Grenoble, F), ID31 beamline (now moved to ID22), using the standard capillary sample holder of the Debye–Scherrer geometry with a 20 keV (0.063 25 nm) monochromatic beam. The IPF was determined using the same procedure described above for the laboratory XRD data.
III. RESULTS AND DISCUSSION
As shown by the SEM micrographs [see Figures 1(a) and 1(b)], mechanical grinding yields a powder of agglomerate particles, which even after an extensive milling ranges from a few to a few tens of micrometers in size. When observed at high magnification, as shown in Figure 1(c), agglomerates show finer features, about 10–20 nm and smaller, which are likely in the size range of the crystalline domains contributing to the diffraction line profiles. Such an agglomeration is a positive feature for the purpose of producing a stable iron-alloy powder, as the inevitable passivation only affects the surface of the agglomerates, thus drastically limiting the oxidation of the metallic nanocrystalline domains.

Figure 1. (Color online) SEM micrographs of the FeMo alloy powder ball milled for 128 h (courtesy of R. Ciancio, CNR-Tasc, Trieste). Agglomerate particles are shown in Figures 1(a) and 1(b), whereas a high magnification picture 1(c) highlights the nanoscale features visible on the agglomerate surface. See text for details.
Stability of the ball milled FeMo alloy powder is demonstrated by the XRD results. XRD data are shown plotting the intensity versus the reciprocal distance 1/d, defined as 1/d = 2/λ[sin(θ)]. This allows a direct comparison of instrumental resolution for the XRD data collected with different wavelengths. Figure 2 shows a comparison of powder patterns collected as the sample of this study by the same instrument at a distance of 10 years. The two patterns look remarkably similar, even considering that differences in the instrument are also to be expected after 10 years of use, maintenance, and substitution of the X-ray source.

Figure 2. (Color online) Comparison of XRD patterns of the powder of this study, a FeMo alloy ball milled for 128 h, collected by the same instrument at a distance of 10 years (respectively 2004 and present day 2014).
Further evidence on the stability of the ball milled powder is provided by LPA, using the whole-powder pattern modeling (WPPM) approach (Scardiand Leoni, 2002, 2006, Scardi et al., Reference Scardi, Ortolani and Leoni2010). The XRD powder data were modeled assuming spherical crystalline domains with a lognormal distribution of diameters, using lognormal mean and variance as refinable parameters. Microstrain effects were completely attributed to dislocations, described within the framework of the Krivoglaz–Wilkens (KW) theory (Krivoglaz and Ryaboshapka, Reference Krivoglaz and Ryaboshapka1963; Wilkens, Reference Wilkens, Simmons, de Wit and Bullough1970a, Reference Wilkens1970b; Martinez-Garcia et al., Reference Martinez-Garcia, Leoni and Scardi2009) in terms of edge/screw dislocations in the primary slip system of iron; refined parameters were: the average dislocation density (ρ), effective outer cut-off radius (R e), and fraction of edge/screw dislocation types (f E). The unit-cell parameter (a 0) was also refined, whereas the IPF was previously determined on a standard LaB6 powder pattern and then convolved with the line profiles of the size and strain contributions. Details of the procedure are available in the literature on the WPPM method (Scardi and Leoni, Reference Scardi and Leoni2002, Reference Scardi and Leoni2006; Leoni et al., Reference Leoni, Confente and Scardi2006; Scardi et al., Reference Scardi, Ortolani and Leoni2010). Further parameters of WPPM include aberrations in sample position and coefficients of a Chebyshev polynomial background.
WPPM results are shown in the graphical form in Figure 3, whereas Table I reports the statistical indices and the most significant microstructural refinement parameters used in the fitting procedure. Figure 3 also shows the details of instrumental component (FWHM and Lorentzian fraction as a function of 1/d). It is quite evident the higher resolution (i.e., smaller FWHM) of the SR XRD data, which also includes a larger number of peaks than the traditional laboratory XRD data [cf. Figures 3(a)–3(c)].

Figure 3. (Color online) Graphical result of WPPM for the powder data of Figure 2, referred to measurements collected on 2004 (a) and 2014 (b). It is also shown the WPPM of an SR-XRD pattern collected on 2004 (c). IPF data (d): FWHM and Lorentzian profile fraction for the laboratory XRD data in (a) and (b) (dash), and SR XRD in (c) (line).
Table I. WPPM results and statistics for data collected on 2004, 2014, combined fit of both datasets (2004 & 2014), and SR XRD data (2004 SR): unit-cell parameter (a 0), average dislocation density (ρ), effective outer cut-off radius (R e), fraction of edge/screw dislocations (f E), Wilkens parameter Re · √ρ, mean domain size (diameter, 〈D〉) and standard deviation (s.d.), specimen displacement; Weighted sum of squares (WSS), Goodness of Fit (GoF); number of refined parameters (Nfit); and total number of data points (Nobs). Estimated standard deviation (e.s.d.) are given in parentheses, referred to the last significant digit.

aError on the zero of the 2θ axis (in degrees).
The stability of the ball milled powder is quite evident by comparing WPPM results for data collected on 2004 and 2014, and the corresponding result is of a joint refinement of the two datasets (same microstructural parameters for both datasets; specimen displacement from the diffractometer axis, IPF, and background are specific to each dataset). Differences, if present, are within the estimated standard deviations, with the exception of the unit-cell parameter, which tends to be affected by the broadness of the peak profiles, and likely correlates with the instrumental errors (specimen displacement, zero error of the 2θ axis). However, this information is only subsidiary to the most relevant result concerning the line profiles, showing significant broadening from both domain size and microstrain effects. The latter, that was completely attributed to dislocations, treated according to the KW theory, corresponds to a high density of interacting dislocations; in fact, the low value of the Wilkens parameter (
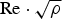 ${\mathop{\rm Re}\nolimits} \cdot \sqrt \rho $
) points out a strong interaction between dislocations, as in dipoles and wall structures. Values of this parameter, being all above unity, also confirm the results that are within the validity limits of the KW dislocation theory (Wilkens, Reference Wilkens, Simmons, de Wit and Bullough1970a, Reference Wilkens1970b).
${\mathop{\rm Re}\nolimits} \cdot \sqrt \rho $
) points out a strong interaction between dislocations, as in dipoles and wall structures. Values of this parameter, being all above unity, also confirm the results that are within the validity limits of the KW dislocation theory (Wilkens, Reference Wilkens, Simmons, de Wit and Bullough1970a, Reference Wilkens1970b).
It is legitimate to doubt the reliability of LPA results when data comprise few peak profiles, as the hkl-dependent features might not be completely captured. The powder patterns discussed so far, collected using CuKα radiation (Figures 2 and 3), include just six peaks of the bcc iron alloy. The same sample was then studied with the SR XRD, using a shorter wavelength (0.063 25 nm), such to include several additional peak profiles. Results, listed in the last column of Table I, agree closely with those from the laboratory XRD data, thus confirming the general validity of the WPPM procedure.
The reliability of the powder and the analysis technique is confirmed by the experimental evidence, which despite differences in the experimental setup (flat plate versus capillary sample holder; parafocusing Bragg–Brentano versus quasi-parallel beam Debye–Scherrer geometry; monochromatic versus two wavelength X-ray beams), shows remarkably similar results. It is however clear that, as expected, using SR provides results that are on an average more accurate than those from a laboratory source. Moreover, it is important to stress that the CuKα wavelength (λ = 0.154 18 nm) used in the laboratory equipment is very close to the K-edge absorption of the Fe contained in the sample (λ = 0.174 33 nm), resulting in a limited penetration depth (~5 μm), affecting the signal intensity of the collected diffraction profile.
The extensive broadening (well beyond the breadth of the instrumental profile), with a substantial contribution of both size and microstrain effects, makes the specimen, ideal for microstructure studies. Coupled with a proven stability in time, this renders the powder as a good candidate for a reference material in LPA studies.
IV. CONCLUSION
Extensive ball milling of a bcc iron-alloy powder yields an ideal candidate as a reference material for LPA. Plasticity effects are such to produce visible and easily measurable broadening of the diffraction peak profiles, caused both by the small size of the crystalline domains and by the strain attributable to lattice defects.
Unless deliberately exposed to highly oxidizing conditions, oxides phases are absent or present in little amount. The powder, in fact, tends to form relatively large agglomerates (tens of micrometers) of much finer crystalline domains; just as a thin passivation layer can form on the agglomerate surface, thus protecting the nanocrystalline domains from further oxidation. Stability was proved over a time range of 10 years.
ACKNOWLEDGEMENTS
The authors thank Dr. Mirco D'Incau (University of Trento) for the preparation of the ball milled sample. The authors acknowledge the European Synchrotron Radiation Facility for the provision of synchrotron radiation facilities and they thank the staff of the ID31 (now ID22) beamline for assistance in data collection.




