


I. INTRODUCTION
The solubility of drugs is a major problem for the pharmaceutical industry because it is a determining factor in their pharmacokinetics, causing differences in the dissolution rate at the sites where their pharmacological action should be exerted. Derivatization of bioactive compounds through their corresponding salt formation is considered to be the most effective and used method to increase the solubility of both acidic and basic pharmaceutical products (Serajuddin, Reference Serajuddin2007) In pharmacology, amines are compounds of high commercial interest because a large number of these have diverse biological activities, including antifungal, antiviral, and antimycotic activities. Secondary amines with naphthalenic rings are of great importance in pharmaceutical chemistry for searching compounds with biological activity (Vargas et al., Reference Vargas, Castelli, Kouznetsov, Urbina, López, Sortino, Enriz, Ribas and Zacchino2003; Kouznetsov et al., Reference Kouznetsov, Vargas, Sortino, Vásquez, Gupta, Freile, Enriz and Zacchino2008). Allylamines based on naphthalene rings, such as terbinafine and naftifine, are good options in the treatment of the diseases caused by fungus (Schäfer-Korting et al., Reference Schäfer-Korting, Schoellmann and Korting2008). Many drugs and biologically important amines are typically used as salts, which are less susceptible to decomposition by oxidation and other reactions, because they are soluble in water and are easily transformed into solutions for use in the form of syrups or injectable solutions (Graham and Fryhte, Reference Graham and Fryhte2011). In previous studies, we reported the results of crystallographic studies of related amine compounds (Camargo et al., Reference Camargo, Henao, Amado and Kouznetsov2010, Reference Camargo, Habran, Henao, Amado and Kouznetsov2011). In this study, we report the synthesis and crystallographic characterization of a new compound 1-N-(3-pyridylmethyl)aminonaphthalene hydrochloride.
II. EXPERIMENTAL
A. Synthesis
The new compound 1-N-(3-pyridylmethyl)aminonaphthalene (4) was prepared by first synthesizing an intermediary imine obtained by reaction of α-naphthylamine (1) and 3-pyridincarboxaldehyde (2) in anhydrous ethanol to obtain the intermediary imine (3) the reduction of which with NaBH4 in anhydrous methanol yields amine (4). This compound was purified with a chromatographic column. Finally, the hydrochloride salt of the organic compound (4) was prepared by addition of hydrochloric acid–ethyl acetate solution in the ratio of 1:3 with constant stirring and maintaining the temperature between 0 and 5 °C for 20 min, obtaining the respective derivative, 1-N-(3-pyridylmethyl)aminonaphthalene hydrochloride (5)
The entire synthesis process is shown in Figure 1, and the title compound was obtained by first reacting 2.00 g of (1) (14 mmol) with 1.79 g of (2) (17 mmol; 1.60 ml) in ethanol (40 ml) and backflowing for 8 h to yield the pure product (3) as yellow oil (2.77 g, 11.92 mmol; 85%), C16H12N2 (MW 232.3 g mol−1). IR: 1624, 1569, 1420, and 774 cm−1. This was followed by reaction of 2.77 g of (3) (11.92 mmol) with 1.35 g of NaBH4 (35.77 mmol) in anhydrous methanol (40 ml), removal of the solvent, and concentration to yield the pure amine (4) as a shine white solid (2.51 g, 10.72 mmol; 90%): m.p. 74–76 °C. C16H14N2 (MW 234.3 g mol−1). IR: 3378, 1583, 1533, 1408, and 768 cm−1. GC–MS (70 eV): t R = 26.63 min, m/z M+ 234(100), 115(60), 142(55), 233(17). 1H-NMR (400 MHz, CDCl3, Me4Si) δ 8.65 (d, J = 1.6 Hz, 1H), 8.50 (dd, J = 4.8, 1.6 Hz, 1H), 7.75 (ddd, J = 20.0, 12.5, 5.5 Hz, 3H), 7.45–7.36 (m, 2H), 7.28–7.20 (m, 3H), 6.52 (dd, J = 7.0, 1.5 Hz, 1H), 4.68 (s, 1H), and 4.48 (s, 2H). 13C-NMR (100 MHz, CDCl3, Me4Si) δ 149.38, 148.94, 142.69, 135.37, 134.63, 134.31, 128.83, 126.52, 125.96, 125.05, 123.71, 123.47, 119.83, 118.28, 105.07, and 46.09. Finally, 0.41 g of (4) (1.75 mmol) was dissolved in ethyl acetate (30 ml) followed by slow addition of the HCl acidified solution to yield (5) as a yellow polycrystalline solid (0.40 g, 1.48 mmol; 84%): m.p. 136–139 °C. C16H15N2Cl (MW 271.77 g mol−1).

Figure 1. Synthesis of 1-N-(3-pyridylmethyl)aminonaphthalene hydrochloride.
B. Powder data collection
A small portion of the title compound was gently ground in an agate mortar and sieved to a grain size less than 37 μm (greater than 400 mesh). The specimen was mounted on a zero-background specimen holder (Buhrke et al., Reference Buhrke, Jenkins and Smith1998). The X-ray powder diffraction (XRPD) pattern was recorded with a D8 FOCUS BRUKER diffractometer operating in Brag-Brentano geometry equipped with a Cu target X-ray tube (40 kV and 40 mA), a nickel filter and a one-dimensional LynxEye detector. A fixed antiscatter slit of 8.0 mm, a receiving slit of 1.0 mm, a soller slit of 2.5°, and a detector slit of 3.0 mm were used.
The scan range was 3–70° 2θ with a step size of 0.02° and a count time of 2 s per step. Powder data were collected at room temperature (298 K).
PowderX analytical software (Dong, Reference Dong1999) was used to remove the background (Sonnerveld and Visser, Reference Sonneveld and Visser1975), to smoothen the experimental XRD pattern (Saviztky and Golay, Reference Saviztky and Golay1964), and finally to eliminate the CuKα 2 component (Rachinger, Reference Rachinger1948). The second derivative method was used to determine the position and intensity of the CuKα 1 diffraction peak of each reflection.
III. RESULTS AND DISCUSSION
The X-ray powder pattern and the XRPD data for the title compound (5) are given in Figure 2 and Table I, respectively. All reflections were indexed successfully using the DICVOL06 program (Boultif and Loüer, Reference Boultif and Loüer2006) on a monoclinic unit cell and the peak positions, each with an absolute error of 0.03° in 2θ, were used in the calculations. The space group, P21/m (No. 11), estimated by the program CHEKCELL (Laugier and Bochu, Reference Laugier and Bochu2002) was compatible with the systematic absences. The unit-cell parameters of the compound (5) were refined with the program NBS*AIDS83 software (Miguell et al., Reference Miguell, Hubbard and Stalick1981). Its crystal data and figures of merit M 20 (Wolff, Reference de Wolff1968) and F 30 (Smith and Snyder, Reference Smith and Snyder1979) are compiled in Table II.
Figure 2. Powder X-ray diffraction pattern of 1-N-(3-pyridylmethyl)aminonaphthalene hydrochloride.
Table I. X-ray powder diffraction data of 1-N-(3-pyridylmethyl)aminonaphthalene hydrochloride. CuK α 1 radiation (λ = 1.5406 Å).
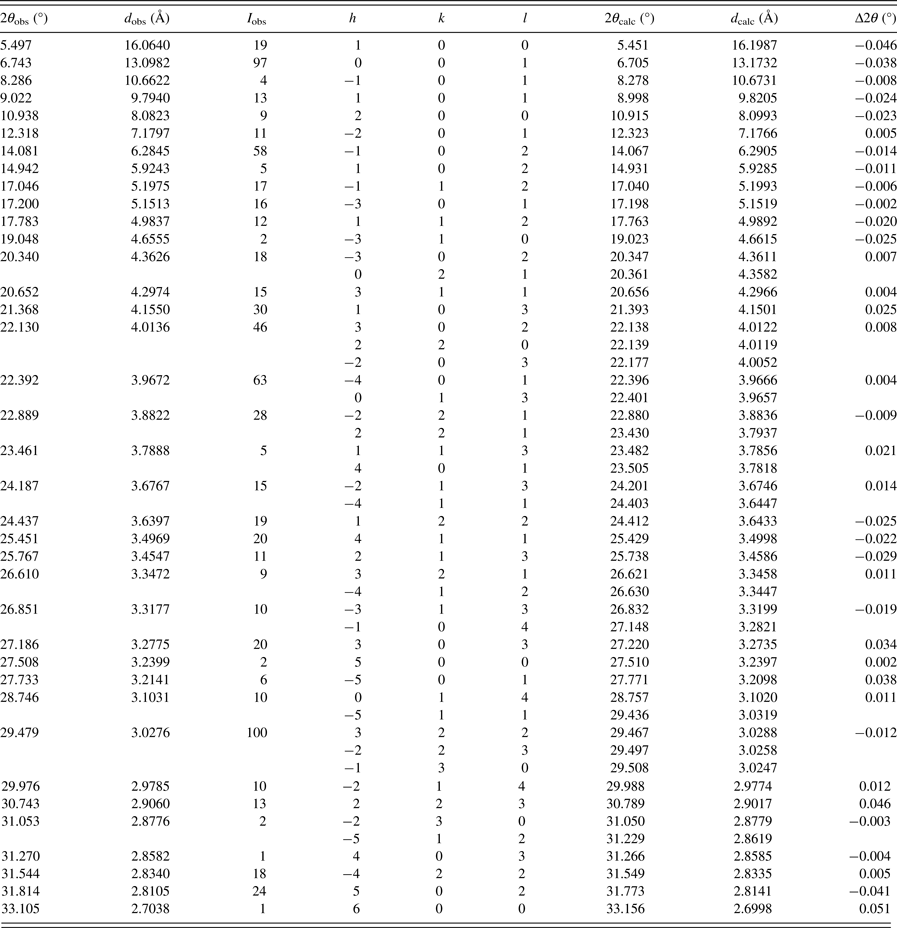
Table II. Parameters obtained by X-ray powder diffraction for 1-N-(3-pyridylmethyl)aminonaphthalene hydrochloride (5).
ACKNOWLEDGEMENTS
This work was supported by Vicerrectoría de Investigación y Extensión – Universidad Industrial de Santander (Project No. 5176). The authors also acknowledge Instituto Zuliano de Investigaciones Científicas, INZIT (Maracaibo-Venezuela) for data collection.




