INTRODUCTION
Calcium hexafluorosilicate bishydrate, CaSiF6·2H2O, CAS Number: 16961-80-1, is a widely marketed chemical, and a precursor of the anhydrous form, CaSiF6, both used as white pigments in ceramics, as preservatives in wood, rubber and textiles, as hardeners in concrete, as opalizers in glass and porcelain enamel and, finally as flotation agents or in insecticides. Medical applications for dentine remediation (Kawasaki et al., Reference Kawasaki, Ishikawa, Suge, Yoshiyama, Asaoka and Ebisu1996) and in bone surgery reconstruction (Hantson et al., Reference Hantson, Mahieu, Gersdorff, Sindic and Lauwerys1995) have also been devised.
Despite its wide use, the crystal structure of CaSiF6·2H2O remains unknown; except for some very limited structural information (unit-cell and experimental d/I list of powder diffraction peaks), in a form that is difficult to retrieve (NBS, 1982). Another occasional citation appeared several years later (Kondrashev and Bogdanov, Reference Kondrashev and Bogdanov1987), in which the bishydrated form of calcium hexafluorosilicate cell was cited on the fly. Even more surprising is the absence of structural information, in the Inorganic Chemistry Structural Database (ICSD), of the purportedly simpler anhydrous CaSiF6 phase, a crystalline material easily obtainable by thermal treatment, above 160 °C, of the hydrated phase (Patil and Secco, Reference Patil and Secco1975).
In order to fill these gaps, we decided to determine the crystal structures of these species, using laboratory powder diffraction data and ab initio structure solution methods. These methods have been used successfully in the recent past by the authors of this paper (Masciocchi et al., Reference Masciocchi, Galli and Sironi2005) and others (David et al., Reference David, Shankland, McCusker and Baerlocher2005), to retrieve the structural features of polycrystalline species of moderately complex structures. A detailed knowledge of structural model at atomic resolution is indeed necessary in order to employ modern quantification techniques based on whole-pattern profile analysis, such as those implemented in widely used computer programs (e.g., QUANTO, Altomare et al., Reference Altomare, Burla, Giacovazzo, Guagliardi, Moliterni, Polidori and Rizzi2001; et al. TOPAS-R, Bruker AXS, 2005).
EXPERIMENTAL
Synthesis of CaSiF6·2H2O
A suspension of SiO2 (particle sizes between 0.063 and 0.200 mm, 70-230 mesh ASTM, 3.30 g) in water (20 ml), was carefully treated with 14.45 ml of HF (40% w/w) at room temperature. To the resulting clear solution, CaCO3 (5.50 g) was added in small portions until no further gas evolution was detected. The suspension was then oven-dried at T = 120 °C for about 150 min, giving a white crystalline powder.
Synthesis of CaSiF6
Anhydrous CaSiF6 was obtained by heating powders of CaSiF6·2H2O in air, at T = 140 °C for about 30 min, in a custom-made furnace (assembled by Officina Elettrotecnica di Tenno, Ponte Arche, Italy) providing in situ heating of the samples directly in the diffractometer cradle.
X-ray powder diffraction characterization
Fine powders, obtained by thoroughly grinding the prepared material in an agate mortar, were deposited on a quartz zero-background plate. Diffraction data were recorded on a Bruker AXS D8 Advance diffractometer operating in the θ:θ mode, equipped with a Lynxeye position-sensitive

Figure 1. (Color online) Rietveld Refinement plot for CaSiF6.2H2O with difference plot and peak markers at the bottom. Horizontal axis 2θ(°); vertical axis: counts. The insert shows the high-angle region at a magnified scale (5×).
detector and Ni filtered Cu Kα radiation (λ = 1.5418 Å). The X-ray generator and diffractometer settings were: 40 kV, 40 mA, and DS 0.5°. The experimental conditions were: step scan mode, with 5 ≤ 2θ ≤ 105°, Δ2θ = 0.02°, total time (in recycling mode) = 18 h. Silicon NBS 640b was used as an external standard.
The recorded pattern of CaSiF6·2H2O is in agreement with that reported in the PDF 00-33-0307 (ICDD, Reference McClune2005), for which a tentative unit cell (but not a structural model) was proposed (NBS, 1982). Peak search methods and indexing of the first 20 lines (2θ < 31 °) by TOPAS-R (Bruker AXS, 2005) allowed the preliminary determination of a monoclinic structure with approximate unit-cell parameters a = 10.48 Å, b = 9.18 Å, c = 5.74 Å, b = 98.95°, V = 545 Å3, and GoF = 119.8. A survey of the 2011 version of the ICSD showed an absence of this phase. It was, therefore, decided to solve the structure by ab initio XRPD methods using the experimental data collected as described above. Systematic absences indicated a probable space group, P21/n, later confirmed by successful structure solution and refinement. The structural model employed in the final whole-pattern Rietveld refinement was determined ab initio using simulated annealing (Kirkpatrick et al., Reference Kirkpatrick, Gelatt and Vecchi1983) as implemented in TOPAS-R (Coelho, Reference Coelho2000). According to this real space method, many “tentative” structural models are randomly generated (by positioning a set of atoms or rigid fragments, randomly, within the crystal lattice) and optimized by a combined global optimization algorithm, minimizing the differences between the calculated and the measured diffraction pattern (Rwp) and the “potential energy” of the system (as defined by a set of user-defined, properly weighted, penalty functions addressing the “correct” stereochemistry). The magnitude of the randomization is related to the so-called temperature of the process, while solutions are accepted and ranked according to Rwp only. In our final model, one independent Ca2+ ion, two water molecules (including hydrogen atoms in idealized positions) and independent silicon and fluorine atoms (with soft-restrained Si-F distances) were located in the asymmetric unit, all eventually found to lie in general positions. The final refinement showed evidence of a small contribution from a contaminant phase, identified as nanocrystalline CaF2 (with crystal size near 40 nm), whose contribution was included in the total pattern simulation—using the known fluorite structural parameters—resulting in a weight percent fraction near 8%.

Figure 2. (Color online) Schematic drawing of the crystal packing of the CaSiF6·2H2O species. Calcium and silicon atoms drawn as large spheres; Calcium ions connected to hexafluorosilicates by fragmented lines. (a) View down [100]. Vertical axis: c. Relevant bond distances (Å) and angles (°): Ca-O1 2.453(4) to 2.554(6) Å, Ca-O2 2.404(5), Ca-O1-Ca 108.8(2); Ca-F (fragmented lines): range: 2.235(3)- to 2.444(3), average: 2.356 (statistical dispersion value 0.076); Si-F 1.648(4) to 1.701(3), average value: 1.672. (b) View down [001]. Vertical axis: a. Fragmented lines indicate OH…O and OH…F hydrogen bonds interactions (discussed in the text).

Figure 3. (Color online) Rietveld Refinement plot for CaSiF6 with difference plot and peak markers at the bottom. Horizontal axis: 2θ (°); vertical axis: counts. The insert shows the high-angle region at a magnified scale (5×).
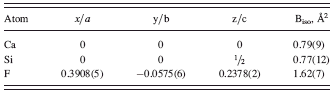
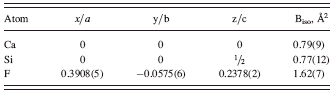
The pattern of anhydrous CaSiF6, apart from the expected contamination of residual fluorite (see above), gave broad(er) peaks, which, nevertheless, allowed an easy indexation [GOF(18) = 34.7] and the determination of trigonal unit-cell parameters (R-centered lattice), later refined by the Le Bail method as a = 5.349 and c = 13.596 Å. A quick search in the ICSD allowed the detection of a number of isomorphous species (among which germanates, stannates, and plumbates), from which initial coordinates of the crystallographically independent ions were taken: Ca in 0, 0, 0; Si in 0, 0, ½ and F in a general (x, y, z) position.
In the final steps of the Rietveld refinements, again performed by TOPAS-R, the background contribution was modelled by a polynomial function, a preferred orientation was applied (only for CaSiF6·2H2O—in the second-order spherical harmonics description approximation), and three different isotropic thermal parameters were separately refined for the Ca, Si and F/O/H atoms. Ionic scattering factors were used for Ca2+, Si4+, and F− ions, while neutral ones were employed for the (more) covalently bound O and H atoms. Peak shapes were described by the fundamental parameters approach with anisotropic peak broadening. These specimen-dependent effects, following a 1/cos θ trend, are attributed to small crystallite sizes, or, more specifically, small coherent scattering domains. Comparison of the average peak widths before and after water molecules elimination allowed the detection of the deterioration of crystallinity, which is attributed to crystal fragmentation. The final Rietveld refinement plots and a sketch of the crystal structures are shown in Figures 1–4. Table I contains the relevant crystal data and data analysis parameters, and Tables II and III contain the final fractional atomic coordinates. While for CaSiF6·2H2O a suitable entry is present in the PDF 00-33-0307 (ICDD, Reference McClune2005), the absence of powder diffraction data for CaSiF6 requires database deposition of the contents of Table IV, in which the observed peaks, in terms of d-spacings, 2θ-angles, relative intensities and their pertinent hkl indices, are listed.
DISCUSSION
The crystal structure of CaSiF6·2H2O contains Ca2+ ions in square antiprismatic coordination, connected to five fluorine atoms from neighbouring SiF62- anions, and three oxygen atoms from coordinated water molecules, two of the bridging type (Ca-O1 2.453(4) to 2.554(6) Å, Ca-O1-Ca 108.8(2)°) and one terminally bound (Ca-O2 2.404(5) Å). CaSiF6·2H2O is, therefore, isomorphous with its heavier analogue, SrSiF6·2H2O, the structure of which was determined from conventional single-crystal methods by Golovastikov and Belov (Reference Golovastikov and Belov1982). In the latter species, six crystallographically independent Si-F bond distances were determined, averaging 1.686 Å, with a very narrow statistical distribution (0.008 Å). Such observations strengthen our choice in using a restrained description of the complex anion in CaSiF6·2H2O, which gave a slightly larger (0.018 Å) statistical spread of Si-F distances (range: 1.648(4) to 1.701(3) Å, average value: 1.672 Å), likely dependent on the inherent limitations of the powder vs single-crystal, diffraction method. As expected, the presence, in SrSiF6·2H2O, of a slightly larger cation than in the calcium analogue (rVIII 1.26 vs 1.12 Å, respectively, Shannon and Prewitt, Reference Shannon and Prewitt1969), inflates all pertinent values, locally (see for example, Sr-O 2.65 to 2.68 Å, Sr-O 2.59 Å), or globally (unit-cell parameters are inflated by ca. 2.6%, the maximum increase (>3.0%) being observed along c).
TABLE I. Crystal data and data-analysis parameters for CaSiF6·2H2O and CaSiF6.

a Profile agreement factors from conventional Rietveld refinement, as defined in TOPAS-R (Bruker AXS, 2005), have been calculated without background subtraction.

Figure 4. (Color online) Schematic drawing of the crystal packing of the CaSiF6 species, viewed down [100]. Vertical axis: c. Color codes: calcium: yellow; silicon: grey; fluorine: green. Relevant bond distances (Å): Ca-F (fragmented lines) 2.269(3); Si-F 1.659(3) Å.
The analysis of the short O···O and O···F contacts in the structure allowed to uniquely define the network of hydrogen bonds further connecting the different units in a complex manner (see Figure 2(b)); with hydrogen atoms positioned in ideal locations at 0.90 Å form the pertinent O atoms, the following contacts can be devised: O1-H11···O2 2.78 Å, O1-H12···F4 2.76 Å, O2-H22···F4 2.77 Å, while H21 belongs to a more complex, bifurcated O2-H21-(F3,F4) interaction.
Thermodiffractometric experiments were performed in order to determine the linear thermal expansion coefficients and to verify the solid-to-solid transformation of the bishydrated phase into CaSiF6. The refined unit-cell parameters, corrected for specimen-displacement errors, showed unexpected variations (see Figure 5), with a negative trend for a (∂lna/∂T = −2.8 × 10−6 K−1) and positive values for b and c (∂lnb/∂T = 7.9 × 10−6; ∂lnc/∂T = 8.4 × 10−6 K−1),
TABLE II. Fractional atomic coordinates for CaSiF6·2H2O.

TABLE III. Fractional atomic coordinates for CaSiF6.
resulting in an overall (rather small) volumetric coefficient of ∂lnV/∂T = 14.1 × 10−6 K−1. Why the structure tends to shrink along a, upon mild heating, is not evident, even after a detailed analysis of the Ca2+ coordination geometry and of the subtle intermolecular contacts. In the absence of further experimental evidence, we tentatively put forward the hypothesis that a slight tilting (reorientation) of the rather stiff hexafluorosilicate anion, modifying the (flexible) octacoordination of the Ca2+ (not possessing stereochemically active d electrons) to a very limited extent, can indeed be invoked for such an unexpected (but small) effect.
The CaSiF6 phase was prepared upon heating the bishydrated species in air at 140 °C for about 30 min. Interestingly, this transformation occurred at a significantly lower temperature than the values of 160 to 165 °C reported by Patil and Secco (Reference Patil and Secco1975) and by Zachara and Wiśniewski (Reference Zachara and Wiśniewski1995), and showed that fragmentation of the pristine polycrystalline material occurred, down to coherent domain sizes of less than 40 nm (as measured by the lorentzian peak
TABLE IV. List of observed diffraction peaks for CaSiF6 (λ = 1.5406 Å).

a A few larger than usual Δ2θ values appear in this table, since several peaks are (i) partially overlapped and (ii) significantly broadened by limited coherent domain sizes, lower than 40 nm (see text).

Figure 5. (Color online) Relative Δx/x (=Δlnx) variations of unit-cell parameters and volume, vs. T (°C). Derived linear thermal expansion coefficients are: ∂lna/∂T = −2.8 × 10−6; ∂lnb/∂T= 7.9 × 10−6; ∂lnc/∂T = 8.4 × 10−6; and ∂lnV/∂T = 14.1 × 10−6 K−1.
broadening attributed to isotropic crystal-size effects after corrected for the instrumental contribution by the fundamental parameters approach). The refined structure of the highly symmetric CaSiF6 phase allowed an independent determination of Si-F bond distances [1.659(3) Å, 6×], only marginally lower the average value cited above for CaSiF6·2H2O, 1.672 Å.
CONCLUSION
The crystal structures of CaSiF6·2H2O and CaSiF6, two species for which single crystals amenable to conventional diffraction techniques were not available, were determined by employing laboratory X-ray powder diffraction data and, for the bishydrated phase, state-of-the-art ab initio techniques. The final model of CaSiF6·2H2O was eventually refined, in P21/n, by the Rietveld method. At variance, the structure of highly symmetric anhydrous salt was refined starting from the knowledge of isomorphous species found in the ICSD database. Accordingly, the detailed knowledge of their crystal structures makes it now possible to meaningfully apply quantitative analytical methods based on the Rietveld technique in the characterization of polyphasic mixtures containing one or both crystal phases, bearing significant industrial interest.
ACKNOWLEDGMENTS
The authors thank Dr. Angelo Maspero (University of Insubria) for helpful discussions. S.F. gratefully acknowledges Dr. Alberto Biavati (Bormioli Rocco) for fruitful suggestions on hexafluorosilicates chemistry.