


I. INTRODUCTION
The topic of crystal structure solution from powders is still young (Le Bail et al., Reference Le Bail, Cranswick, Adil, Altomare, Avdeev, Cerny, Cuocci, Giacovazzo, Halasz, Lapidus, Louwen, Moliterni, Palatinus, Rizzi, Schilder, Stephens, Stone and van Mechelen2009). The direct-space methods for crystal structure determination from powder diffraction have been validated by academic and industrial laboratories around the world, bringing a new capability to the powder diffraction technique. However, generally, crystal structures from powders are less accurate and often less “credible” than the outcome of single-crystal diffraction analysis. Several contributing factors can be mentioned: the projection of the three-dimensional reciprocal lattice into one dimension in the powder diffraction pattern, and the consequent loss of independently measurable diffraction intensities; the possible error correlation in Rietveld parameters obscuring model deficiencies; the limited experience of the scientific community, the lack of automation, and considerable need of human intervention in powder data analysis; the lack of an absolute scale to distinguish between good and poor Rietveld agreement factors (R wp, χ 2, R I, R p); and consequently, the partially subjective assessment of the model validity (Toby, Reference Toby2006).
Although in the general case, crystal structure determinations by single-crystal diffraction render coordinates closer to the true values than those obtained from powders, advances in instrumentation, algorithms, and software make nowadays possible to tackle structural problems from powders, which have been simply not solvable during most of the 20th century, opening up new possibilities for the revision and improvement of early structural models. This article describes such case revealed through the redetermination of the crystal structure of [Co(NH3)5Cl]Cl2, the “purpureo salt,” previously reported from X-ray single-crystal diffraction in the space group Pnma (No. 62) (Shigeta et al., Reference Shigeta, Komiyama and Kuroya1963; Messmer and Amma, Reference Messmer and Amma1968). The analysis of its modern synchrotron X-ray powder diffraction data indicates that the space group is Pn21 a (No. 33). The crystal structure results are discussed, the three models are compared, and the powder results are supported by the Rietveld agreement factors corresponding to the single-crystal and powder structures, as well as by the observation of the powder crystallites under the microscope to discard considerable preferred orientation effects. In addition, a search in the Cambridge Structural Database (Allen, Reference Allen2002) was used to determine the validity of the bond distances and overall coordination complex geometry derived from powders.
II. EXPERIMENTAL
A. X-ray powder diffraction
The synchrotron X-ray powder diffraction data of [Co(NH3)5Cl]Cl2 was collected at the X16C beamline of the N.S.L.S.-I, Brookhaven National Laboratory, USA. The pink [Co(NH3)5Cl]Cl2 powder was loaded into a 0.7 mm diameter glass capillary, and the data were collected at room temperature using a capillary spinner. The wavelength 0.700 12 Å was selected with a Si(111) double monochromator. The wavelength calibration was carried out using an Al2O3 NIST standard. The incident parallel beam was monitored with an ion chamber, and a Ge(111) analyzer crystal was placed after the sample and before the detector to increase the angular resolution, whereas the out-of-plane resolution was given by slits. A NaI(Tl) scintillation detector was used to measure the diffracted radiation.
B. Crystal structure determination from powders
The X-ray powder diffraction data of [Co(NH3)5Cl]Cl2 was indexed with TREOR (Werner et al., Reference Werner, Erikson and Westdahl1985) leading to the unit-cell parameters a = 13.287(5) Å, b = 10.34(1) Å, c = 6.718(3) Å, α = β = γ = 90°, V = 923.39 Å3 (M 20 = 29); and with DICVOL06 (Boultif and Louër, Reference Boultif and Louër2004) leading to the lattice parameters a = 13.2704 Å, b = 10.3300 Å, c = 6.7098 Å, α = β = γ = 90°, V = 919.81 Å3 (M 20 = 43.2). The unit-cell parameters were refined through a Le Bail fit (Le Bail, Reference Le Bail2005) in the space group P222 (No. 16) using GSAS (Larson and von Dreele, Reference Larson and vonDreele2004). Those values (a = 13.2665 Å, b = 10.3382 Å, c = 6.7087 Å, α = β = γ = 90°) were used for crystal structure determination. The Rietveld graph is shown in online Supplementary Figure S1, and the agreement factors were R wp = 5.07%, χ 2 = 2.26, and R p = 3.98%.
The crystal structure was solved using direct methods as implemented in the software EXPO2013 (Altomare et al., Reference Altomare, Cuocci, Giacovazzo, Moliterni, Rizzi, Corriero and Falcicchio2013), which also indexed the powder diffraction data, yielding basically the same unit-cell parameters, and it suggested two space groups. The largest figure of merit was 0.196 for Pn21 a (No. 33), followed by 0.113 for Pnma (No. 62).
The Rietveld refinement of the crystal structure model in Pn21 a was carried out with the software GSAS (Larson and von Dreele, Reference Larson and vonDreele2004). The atomic positions of the hydrogen atoms were calculated with SHELX (Sheldrick, Reference Sheldrick2008) as implemented through WinGX (Farrugia, Reference Farrugia2012) and refined with soft bond-length and bond-angle restraints. The following parameters were refined in the Rietveld fits: scale factor, 36 cosine Fourier series coefficients for the background function No. 2, unit-cell parameters, 2θ zero-point error, fractional atomic coordinates, isotropic atomic displacement parameters, and asymmetry-corrected Pseudo-Voigt profile parameters (profile function No. 3). A constant term for X-ray absorption correction suitable for capillary transmission geometry, as implemented in GSAS (Larson and von Dreele, Reference Larson and vonDreele2004), was used. Its value was calculated to be 0.636 Å−1, assuming an absorption linear coefficient of 25.44 cm−1, 0.035 cm capillary radius, 0.5 powder packing coefficient, and 0.700 12 Å radiation.
The scattering factors used were those of Co+3, Cl−, N, and H. The atomic displacement parameter of the hydrogen atoms was constrained to 1.2 times the value of that of the non-hydrogen atoms, both of which were refined subjected to a group constraint. The Rietveld-refined atomic coordinates were corrected following the procedure published in Scott (Reference Scott1983).
III. RESULTS AND DISCUSSION
The crystallographic parameters describing the crystal structure of [Co(NH3)5Cl]Cl2 after Rietveld refinement, and the Rietveld agreement factors are summarized in Tables I and II. Figure 1 shows the coordination sphere around the Co(III) ions and the Co(III)—ligand interatomic distances. Figure 2 shows the Rietveld refinement graph.
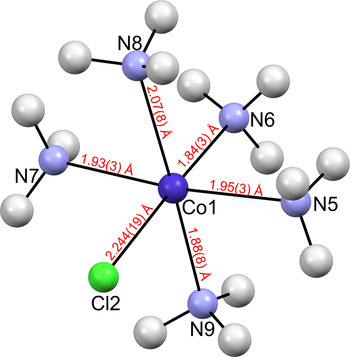
Figure 1. (Color online) The coordination sphere around the Co(III) ions in [Co(NH3)5Cl]Cl2 showing the atom-labeling scheme and selected interatomic distances. Thermal ellipsoids are represented at the 50% probability level. Co3+ ions are shown in dark blue, N in light blue, Cl− in green, and H in light gray.

Figure 2. (Color online) Observed (blue circles), calculated (solid red line), and difference (bottom black line) X-ray powder diffraction patterns of [Co(NH3)5Cl]Cl2 refined by the Rietveld method in the space group Pn21 a (No. 33), orthorhombic. Vertical bar symbols represent allowed peak positions.
Table I. Crystal data of [Co(NH3)5Cl]Cl2 from synchrotron X-ray powder diffraction [space group Pn21 a (No. 33), orthorhombic (standard setting Pna21)].

Table II. Atom labels, fractional atomic coordinates, occupancy factors, and isotropic atomic displacement parameters of [Co(NH3)5Cl]Cl2 from the analysis of synchrotron X-ray powder diffraction data.

It has been pointed out (Toby, Reference Toby2006) that the best way to judge the correctness of a model from powders is: (1) graphically and carefully observing the experimental and calculated Rietveld profiles; and (2) confirming that the chemical bonding in the model is plausible. For this material, since powder and single-crystal diffraction give rise to crystal structure descriptions in two space groups, Pnma and Pn21 a, respectively, and unresolved subtleties in the internal geometry of the coordination complex still remained after the work of Messmer and Amma (Reference Messmer and Amma1968); the Rietveld method was also used to further assess the validity of the published models from single-crystal diffraction (Shigeta et al., Reference Shigeta, Komiyama and Kuroya1963; Messmer and Amma, Reference Messmer and Amma1968), comparing their Rietveld agreement factors and Rietveld graphs with those of the model from powders.
The unit-cell parameters reported in 1963 and 1968 are similar to those found from powders (after Rietveld refinement), and the values from the three models are compared in Table III. The unit-cell parameters determined in 1968 are the closest to those calculated from the powder pattern.
Table III. Space group symmetry and unit-cell parameters from the powder model, and the single-crystal models of Shigeta et al. (Reference Shigeta, Komiyama and Kuroya1963) and Messmer and Amma (Reference Messmer and Amma1968).

In order to first assess the validity of the model of Shigeta et al. (Reference Shigeta, Komiyama and Kuroya1963), the unit-cell parameters, 2θ zero-error, background coefficients, and peak profile parameters were refined in a model biased Le Bail fit replacing the space group P222 by Pnma, typing the published atomic coordinates and re-adjusting the L i,j coefficients of the profile function No. 3 (Larson and von Dreele, Reference Larson and vonDreele2004).
In the Rietveld fit that followed, only the scale factor and the background coefficients were initially refined. The calculated pattern showed serious misfits for the most intense peaks and many others. After refining the published atomic coordinates (without bond-length restraints), and the isotropic atomic displacement parameters (subjected to a group constraint), the best agreement factors achieved were R wp = 10.24%, χ 2 = 9.22, R I = 5.78%, R p = 7.67% (in all cases significantly larger than those of our model from powders); and although diminished, the misfits persisted (see online Supplementary Figure S2). Moreover, the two nitrogen positions above and below the mirror plane in the unrefined single-crystal model (Shigeta et al., Reference Shigeta, Komiyama and Kuroya1963) split into four, rendering the refined model implausible. The Fourier difference map showed minimum and maximum values of −0.63 and +0.86 e−/Å3, respectively.
We point out that various factors could have contributed to an erroneous structure (with R = 0.109) reported by Shigeta et al. (Reference Shigeta, Komiyama and Kuroya1963), such as the visual determination of the reflection intensities, the lack of absorption correction, and that the material was simply assumed to be isostructural with a rhodium (III) salt. Individual atomic displacement parameters for each atomic coordinate and hydrogen positions were not reported either.
Towards facilitating the comparison of all three models and their Rietveld refinements, Figure 3 shows the crystal packing in the three models, and Figure 4 shows the reported coordination complex geometry using the labeling scheme of the respective publications (Shigeta et al., Reference Shigeta, Komiyama and Kuroya1963; Messmer and Amma, Reference Messmer and Amma1968). Table IV summarizes the Rietveld agreement factors, Fourier difference residuals, and the number of parameters refined in each fit, all of which were carried out using our synchrotron powder diffraction data (this work).

Figure 3. (Color online) The crystal structure of [Co(NH3)5Cl]Cl2 approximately along the c-axis, with inversion centers shown in orange, 21 screw axes in green, and mirror planes in cyan. Glide planes are not shown for clarity; (a) the model of Shigeta et al. (Reference Shigeta, Komiyama and Kuroya1963) in Pnma, (b) the model of Messmer and Amma (Reference Messmer and Amma1968) also in Pnma, and c) the model obtained from powder diffraction in Pn21 a (H atoms are not shown for clarity). Co3+ ions are shown in dark blue, N in light blue, Cl− in green. The atom labeling shown is that of the original publications (Shigeta et al., Reference Shigeta, Komiyama and Kuroya1963; Messmer and Amma, Reference Messmer and Amma1968). Selected hydrogen bonding is indicated with dashed gray lines and the distances are in Å.

Figure 4. (Color online) The internal geometry of the coordination complex [Co(NH3)5Cl]2+ with reported interatomic distances in Å and the original labeling scheme in the publications cited. (a) The model of Shigeta et al. (Reference Shigeta, Komiyama and Kuroya1963) in Pnma, (b) the model of Messmer and Amma (Reference Messmer and Amma1968) also in Pnma, and (c) the model obtained from powder diffraction in Pn21 a (this work). Co3+ ions are shown in dark blue, N in light blue, Cl− in green, and H in light gray (only reported for the model in Pn21 a).
Table IV. Summary of the equally weighted Le Bail fit in P222 and Rietveld refinement agreement factors for the model from powders in Pn21 a (this work), and the single-crystal models of Shigeta et al. (Reference Shigeta, Komiyama and Kuroya1963) and Messmer and Amma (Reference Messmer and Amma1968) [the latter was refined without and with calculated hydrogen positions; N r is the number of restraints and N Cons is the number of constraints; N Crys is the number of crystallographic parameters, N P the number of profile parameters, and N T the total number of parameters refined, as defined in Scott (Reference Scott1983)].

The number of restraints only refers to the bond-distance and -angle restraints to refine the positions of hydrogen atoms. Those were included in the powder model (neither single-crystal model reported H atoms), and in the Rietveld-refined model of Messmer and Amma (Reference Messmer and Amma1968), for comparison purposes. The number of constraints is one in all cases, and it corresponds to the isotropic atomic displacement parameters of non-H atoms and H atoms (when included).
Following the procedure described in Scott (Reference Scott1983) for the estimation of standard uncertainties from Rietveld fits, the number of crystallographic parameters (N C) is the sum of the three lattice parameters, the number of refinable atomic coordinates, and the atomic displacement parameter. The number of profile parameters (N P) is the sum of 36 background coefficients, the scale factor, the zero-point error, and peak profile parameters (when refined in the last GSAS cycles). The total number of parameters refined (N T) is the sum of N C and N P. The entries in Table IV corresponding to the model from powders in Pn21 a were used in the calculation of the standard uncertainties following the procedure described in Scott (Reference Scott1983).
Furthermore, all three structural models are credible from the chemical-bonding point of view; however, the models describe subtleties in the geometry of the [Co(NH3)5Cl]2+ coordination complex differently. The overall crystal packing in both single-crystal and powder structures is quite similar. The coordination sphere around the Co(III) ion is octahedral, and it is composed of five ammonia molecules and one chloride ion. Viewed approximately along the c-axis, the structures show two alternating horizontal layers, one layer is made of chloride ions, and the second of penta-ammonium cobalt(III) chloride complex ions [Co(NH3)5Cl]2+, respectively. This is represented in Figure 3 for the single-crystal (Shigeta et al., Reference Shigeta, Komiyama and Kuroya1963; Messmer and Amma, Reference Messmer and Amma1968) and powder structures.
However, both single-crystal models have been defined differently. In both cases, Co3+ and three N atoms sit on a mirror plane at y = 1/4, in a 4c special position (x, ¼, z), thus conferring partial planar symmetry to the coordination complex ion [Co(NH3)5Cl]2+ (see Figure 3). In the model of Shigeta et al. (Reference Shigeta, Komiyama and Kuroya1963), described with eight atomic coordinates and constructed by comparison with the structure of the analogous rhodium salt, N4 and N5 (above and below such mirror plane, respectively) are in 8d general positions [see Figure 3(a)]; while in the model of Messmer and Amma (Reference Messmer and Amma1968), described with seven atomic coordinates derived from a Patterson map, the N3 atom above the mirror plane in a 8d (x, y, z) general position generates N3′ (x, y−½, z) below the mirror plane, within the same complex ion [see Figure 3(b)]. The atomic positions of both models above are summarized in Tables S.I and S.II (online Supplementary material).
Moreover, in both single-crystal structures, the layers of chloride ions are generated by only one crystallographically independent chloride. In the powder structure in Pn21 a, the chloride layers are made of two crystallographically independent chloride ions (and a third chloride is coordinated to the cobalt ion). Pn21 a does not have inversion symmetry or mirror planes [see Figure 3(c)], and the [Co(NH3)5Cl]2+ coordination complex does not show sections with planar symmetry.
In order to gather additional information to qualitatively assess the likelihood of observing planar symmetry within the coordination complex, and the credibility of the bond distances resulting from the three models, a search in the Cambridge Structural Database (Allen, Reference Allen2002) version 5.38, November 2016, was conducted to identify minimum, maximum, and average cobalt–NH3 distances, for structures in which cobalt (this means Co2+ or Co3+ ions) was bonded to five ammonia molecules and one other residue of any kind. For R < 5%, 69 entries were found resulting in 345 bond-distance observations. Those varied from 1.834 to 2.188 Å, and the average value was (1.966 ± 0.002) Å. Thus, in principle, the bond lengths shown in Figure 1 are deemed valid. Additionally, an analogous planar symmetry section of the coordination complex imposed by the crystal symmetry was not found in any of the above crystal structures. For the four entries hexa-ammine-cobalt(III) dichloride 3-carboxyprop-2-ynoate monohydrate (ASAWUM); (4-methylimidazole-N)-penta-ammine-cobalt(III) trichloride dihydrate (DUSDAW); hexa-ammine-cobalt(III) tris(methanesulfonate) (WEXRIB); and bis[(hexa-ammine)-cobalt(III)] trioxalate tetrahydrate (XEDNAV), in the space groups Pnma, Pna21, Pnnm, and Pnnm, respectively, only two of the six ligands in the coordination complex were placed on mirror planes for the three above cases in Pnma and Pnnm. Thus, if the space group of the purpureo salt is actually Pnma, it would be the first structure (after the 69 published) wherein four ligands in [Co(NH3)5 R]2+ (for R = any residue) are on a mirror plane.
Figure 4 shows the coordination complex geometry reported in the three models, also using the labeling scheme from the publications cited. Messmer and Amma (Reference Messmer and Amma1968) had identified two possible space groups, Pnma and Pn21 a; however, they chose to report the structure in Pnma because of the observed correlation between the nitrogen atomic positions above and below the mirror plane (N3 and N3′) in Pn21 a refinements, which are symmetry-related in Pnma but crystallographically independent in Pn21 a.
One of the reasons for which this structure was revisited by Messmer and Amma (Reference Messmer and Amma1968) was to further investigate the change in bond lengths that occur in metal complexes of this type resulting from trans-directing ligands, such as the electronegative chlorine atom. In the structural description of Shigeta et al. (Reference Shigeta, Komiyama and Kuroya1963), the Co–N1 distance trans to the Co–Cl bond [see Figures 3(a) and 4(a)] is significantly shorter than the four remaining Co–N distances; this is 1.91 vs. 1.97 (×3) and 1.98 Å. On this issue, Messmer and Amma (Reference Messmer and Amma1968) concluded that a significant trans distortion was not observed within the limitations of their data; however, it was confidently determined that one of the Co–N distances [Co–N(4)] was larger than the remaining four; this is 1.998(6) vs. 1.962(4) (×2), 1.946(6), and 1.987(6) Å [see Figures 3(b) and 4(b)]; and that such N atom is likely involved in hydrogen bonding to a chlorine atom [Cl(2)] [see Figure 3(b)], and this likely gives rise to the longest distance observed for Co–N(4), and the shortest N(4)–Cl(2) distance of 3.333(5) Å, among all N–Cl distances.
Thus, three questions to answer using our synchrotron X-ray powder diffraction data have been identified in our work: (1) Is the space group Pnma correct, or the crystal structure is a pseudosymmetry case and it is better described in Pn21 a?; (2) Is there a Co–N distance significantly larger than the remaining four, such as Messmer and Amma (Reference Messmer and Amma1968) reported?; and (3) Can a significant trans effect be observed in this compound such as Shigeta et al. (Reference Shigeta, Komiyama and Kuroya1963) did, within the limitations of our powder diffraction data?
Toward answering these questions and further compare the models, we also Rietveld-refined the results of Messmer and Amma (Reference Messmer and Amma1968) using our synchrotron powder pattern. The Rietveld fit obtained is shown in online Supplementary Figure S3, and the agreement factors and other Rietveld refinement details are summarized in Table IV. In this case, the Rietveld refinement renders a chemically plausible model; however, the graphical observation of the Rietveld fit again points to serious model deficiencies (see online Supplementary Figure S3).
One could think that considerable preferred orientation effects could explain why the calculated intensities of a few low-angle peaks [this is (011) around 2θ = 7.129° and (210) at 2θ = 7.186°] are below their observed values. Thus, we observed the powder crystallites under the microscope. The micrographs are shown in Figure 5, and they indicate that the powder crystallites are very small (estimated in the order of 1–10 µm) and of a rather spherical shape; thus, preferred orientation corrections are unlikely to be required.

Figure 5. (Color online) Micrographs of the powder crystallites in the [Co(NH3)5Cl]Cl2 specimen used for X-ray powder diffraction data collection. (a) View at 1 µm resolution showing the absence of large needles or plates, and small microcrystallites (estimated 1–10 µm size); (b) one microcrystallite.
To further test the model of Messmer and Amma (Reference Messmer and Amma1968), the hydrogen positions were added with SHELX (Sheldrick, Reference Sheldrick2008) as implemented through WinGX (Farrugia, Reference Farrugia2012), leading to a considerable improvement of the Rietveld fit (online Figure S4); however, the agreement factors (shown in Table IV) remained higher than those obtained in Pn21 a. We must note that Messmer and Amma (Reference Messmer and Amma1968) did not report hydrogen positions, and absorption corrections were not carried out.
Thus, considering all above results, in particular, the lowest agreement factors and Fourier difference residuals obtained for the model from powders in Pn21 a, in principle such model seems physically meaningful to us. We believe the analysis of the synchrotron powder diffraction data rather indicates the validity of the Pnma space group choice reported in Messmer and Amma (Reference Messmer and Amma1968) is questionable, and we note that it has been already an arguable choice in 1968, as the authors mentioned. An additional hint toward the structural description in Pn21 a is given by evaluating the possibilities of formation of energetically favorable hydrogen-bonding motifs in the crystal packing adopted. It seems reasonable that the partial planarity of the [Co(NH3)5Cl]2+ coordination complex, as well as bond distances and angles in the Pnma description, can be slightly distorted because of the bonding energy gains resulting from hydrogen bonding of the −NH groups of ammonia to other nitrogen and chloride ions.
Table V shows the hydrogen-bonding distances calculated by PLATON (Spek, Reference Spek2002) and most of the N–Cl distances leading to hydrogen bonding are also shown in Figure 3(c) (a few were omitted for clarity). It is interesting to note that rather than a considerable deviation from planar symmetry for the atoms Cl2, N5, N6, and N7 [see Figure 3(c)], it rather seems that the angles of N9 and N8 with the above atoms considerably deviate from the (90 ± 1)° values observed for the corresponding N3 and N3′ atoms in Figure 3(b), in Pnma. In Pn21 a, such angles are not related by mirror symmetry, and their values range from 80 to 100° instead. Since these irregular features in the internal geometry of the coordination complex could be explained by the maximization of lattice energy gains by hydrogen bonding within the crystal, we believe that this is a reasonable chemical reason leading to a better description of the structure in Pn21 a, and the structure is a pseudosymmetry case (approximation to a higher symmetry). It must be also mentioned that ADDSYM in the software PLATON (Spek, Reference Spek2002) suggested a space group change to Pnma. However, Spek (Reference Spek2002) indicates that approximations to pseudosymmetry frequently occur in crystal structures; for this reason, the missed symmetry alerts might be false in some cases, and they require careful consideration by the crystallographer analyzing “by hand” the diffraction data.
Table V. Hydrogen-bonding distances (in Å) and angles (in °) calculated by PLATON (Spek, Reference Spek2002) for the crystal structure of [Co(NH3)5Cl]Cl2 from synchrotron X-ray powder diffraction [space group Pn21 a (No. 33), orthorhombic].

Thus, our answer to the first question above is that the comparison of the three models using the modern synchrotron powder pattern, including X-ray absorption corrections, calculated hydrogen atom positions, and discarding preferred orientation effects, indicates the correct space group choice is Pn21 a, not Pnma. This choice is further supported by the possibilities of formation of energetically more favorable hydrogen-bonding motifs in the lower symmetry space group, leading to a pseudosymmetry.
To answer questions 2 and 3, we must analyze in detail the standard uncertainties of the atomic coordinates from powders, keeping also in mind that the hydrogen positions are only calculated since in the general case, powder diffraction data do not afford their experimental determination. Referring to Figure 4(c), we note that the Co–N8 distance, 2.07(8) Å, is larger than the remaining four, 1.84(3), 1.88(8), 1.93(3), and 1.95(3) Å. Thus, using the calculated standard uncertainties and assuming uncorrelated errors, within two standard deviations (95% confidence level), we cannot say from the powder data that this distance is statistically significantly longer than the rest. However, within one standard deviation (68% confidence level), we could affirm it is (additional details are in the online Supplementary material). This result agrees with the analysis of Messmer and Amma (Reference Messmer and Amma1968), who confidently concluded that one of the distances is longer than the other four.
Concerning the trans effect reported in Shigeta et al. (Reference Shigeta, Komiyama and Kuroya1963), and referring again to Figure 4(c), we see that the Co–N6 distance of 1.84(3) Å is indeed the shortest among the five Co–N distances leading to a trans effect. However, through the same analysis and assuming uncorrelated errors, we cannot affirm from our powder data that the Co–N6 distance is the shortest distance among the five, giving rise to a statistically significant trans effect with 68% confidence level (additional details are in the online Supplementary material).
In qualitative terms, we assert that the powder data point to the presence of both effects, but the trans effect is less likely than the one reported by Messmer and Amma (Reference Messmer and Amma1968).
IV. CONCLUSION
This article reports the crystal structure of the purpureo salt, [Co(NH3)5Cl]Cl2, recently re-determined from its synchrotron powder diffraction data in the lower symmetry space group Pn21 a (No. 33). This particular case reminds us that crystal structures solved from single-crystal X-ray diffraction are not always “indisputable.”
Furthermore, a disadvantage of the powder diffraction technique is that there is not an absolute scale to distinguish between good and poor Rietveld agreement factors (R wp, χ 2, R I, R p). However, this work also shows that in addition to carefully evaluating the graphical representation of the Rietveld fit and the overall credibility of the chemical bonding, the comparison of the Rietveld agreement factors of alternative models fitting the same diffraction data, the use of error analysis, the comparison of the crystallographic parameters determined with those of similar structures in the Cambridge Structural Database (Allen, Reference Allen2002), and the observation of the powder crystallites under the microscope are valuable resources to evaluate the validity of models derived from powders.
In summary, we conclude that the crystal structure of the purpureo salt is a pseudosymmetry case, and it should be described using the space group Pn21 a, rather than Pnma. This is likely because of the formation of energetically more favorable hydrogen-bonding motifs in Pn21 a, afforded by a slight distortion of the coordination complex geometry previously reported in Pnma (Messmer and Amma, Reference Messmer and Amma1968).
Additional details of the internal geometry of the [Co(NH3)5Cl]2+ coordination complex have been described. We can affirm that most likely the effect reported in Messmer and Amma (Reference Messmer and Amma1968) (one longest Co–N distance among the five) is real. Furthermore, although probably the trans effect also exists, our confidence level is lower than that the above because of the limitations of our data. To further elucidate these structural details, a modern single-crystal structure determination would be certainly advantageous, although a carefully performed data collection and Rietveld analysis from synchrotron powder diffraction would also lead to such results.
SUPPLEMENTARY MATERIAL
The supplementary material for this article can be found at https://doi.org/10.1017/S0885715617000677.
ACKNOWLEDGEMENTS
This research was in part performed at the National Synchrotron Light Source, Brookhaven National Laboratory, which is supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences (Contract No. DE-AC02- 98CH10886). SP gratefully acknowledges a sample of the purpureo salt from Professor T. Abdel-Fattah at Christopher Newport University, USA, access to the HIROX optical microscope at the surface characterization laboratory of the College of William and Mary, and partial financial support from ICDD (International Centre for Diffraction Data) GIA 08-04.












