


I. INTRODUCTION
Cu2ZnSnS4 (CZTS) has received increased attention in recent years. Its properties, namely a nearly ideal band gap of about 1.5 eV and a high absorption coefficient for visible light (Bacewicz et al., Reference Bacewicz, Antonowicz, Podsiadlo and Schorr2014), make it a promising alternative for absorber layers in thin-film solar cells. In contrast to other candidates, this compound contains only earth abundant and non-toxic elements. Yielding part of this advantage, properties can be further optimized by varying the composition. In particular, Cu2ZnSn(Se,S)4 compounds have already achieved efficiencies above 12% (Barkhouse et al., Reference Barkhouse, Gunawan, Gokmen, Todorov and Mitzi2012; Wang et al., Reference Wang, Winkler, Gunawan, Gokmen, Todorov, Zhu and Mitzi2014). By varying the fractions of copper, zinc, and tin, vacancies and anti-sites can be induced. Moreover, these point defects can likely be controlled by the synthesis conditions (Scragg et al., Reference Scragg, Choubrac, Lafond, Ericson and Platzer-Bjorkman2014).
Despite large numbers of recent publications dealing with CZTS-derived compounds and the apparent importance of the cation distribution for the photovoltaic properties, knowledge about this is still limited. To a large degree this is because of the problems in distinguishing copper and zinc, which of course is necessary in establishing their distribution. Cu1+ and Zn2+ are isoelectronic cations. As a result, their X-ray scattering characteristics, expressed by the atomic form factor f, are nearly identical under conventional circumstances. Standard X-ray diffraction thus cannot distinguish the two elements and can at best provide the amount of vacancies. Methods such as X-ray absorption fine structure (XAFS) (Bacewicz et al., Reference Bacewicz, Antonowicz, Podsiadlo and Schorr2014) or Raman spectroscopy (Scragg et al., Reference Scragg, Choubrac, Lafond, Ericson and Platzer-Bjorkman2014) have been used to analyze the defect structure. However, direct quantification of cation distribution up to now has only been done successfully from neutron powder diffraction (Schorr et al., Reference Schorr, Hoebler and Tovar2007; Schorr, Reference Schorr2011). The neutron scattering lengths of copper and zinc are sufficiently different from each other, which allow the distinction of the two species. Among other things it was settled in these papers that both CZTS and CZTSe compounds crystallize in the kesterite structure, in space group
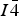 $I{\bar 4}$
. Application of the neutron diffraction is limited by the large amounts of sample typically required, which precludes its application to thin-film samples. Also, long data collection times and the dwindling number of neutron sources worldwide limit this method to “corner stone” problems.
$I{\bar 4}$
. Application of the neutron diffraction is limited by the large amounts of sample typically required, which precludes its application to thin-film samples. Also, long data collection times and the dwindling number of neutron sources worldwide limit this method to “corner stone” problems.
Anomalous diffraction in principle can overcome both problems. More of the synchrotron sources are needed for this method than there are neutron sources, and the high intensity allows using very small sample volumes. As the atomic form factor changes at the absorption edges of the respective elements, Cu1+ and Zn2+ can be distinguished from diffraction data taken with radiation of appropriate energies. Despite this, up to now (Washio et al., Reference Washio, Nozaki, Fukano, Motohiro, Jimbo and Katagiri2011; Nozaki et al., Reference Nozaki, Fukano, Ohta, Seno, Katagiri and Jimbo2012; Lafond et al., Reference Lafond, Choubrac, Guillot-Deudon, Fertey, Evain and Jobic2014) the method has been used only for the qualitative analysis, in particular the confirmation of the kesterite structure instead of the stannite structure. When quantification of the site occupancies was attempted, this turned out to be unexpectedly problematic. Ideally, it should be possible to simultaneously refine multiple datasets with different wavelengths, and from this to directly derive the site occupancy factors for copper and zinc. In practice, the results of this procedure were found to be unstable. The very high pseudosymmetry of the kesterite structure results in strong correlation between the occupancy factors of the different crystallographic sites. The use of powder samples intensifies this, as symmetry-breaking reflections overlap perfectly. Here we demonstrate a recipe, by which stable and reliable results can be derived from powder diffraction patterns.
II. THEORY
A. Correlation of structural parameters
Attempts to refine the occupation factors of copper and zinc simultaneously for the same site, using multiple powder diffraction patterns taken close to the respective absorption edges, failed. The results were found to be very unreliable, with extremely high dependency on the values of other parameters. The least-squares correlation matrix yielded correlation factors up to 99% and higher for actually independent parameters. This can be understood from the particular structural conformation (Figure 1). The kesterite structure in space group
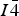 $I\bar 4$
derives by cation ordering from the sphalerite structure in space group
$I\bar 4$
derives by cation ordering from the sphalerite structure in space group
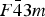 $F\bar 43m$
. As a result the geometry of the four cation sites 2a–2d is very similar. Changes to the structure factor because of changes in the occupation are consequently similar and can be balanced to a large degree by changes in another site. This effect is particularly strong for the pair 2c–2d, which only split at the transition from disordered (
$F\bar 43m$
. As a result the geometry of the four cation sites 2a–2d is very similar. Changes to the structure factor because of changes in the occupation are consequently similar and can be balanced to a large degree by changes in another site. This effect is particularly strong for the pair 2c–2d, which only split at the transition from disordered (
 $I\bar 42m$
) to ordered (
$I\bar 42m$
) to ordered (
 $I\bar 4$
) kesterite. Similarly the coordinates of the anion, xyz with x ≈ y, result in strong correlations between x and y and anion displacement parameters.
$I\bar 4$
) kesterite. Similarly the coordinates of the anion, xyz with x ≈ y, result in strong correlations between x and y and anion displacement parameters.

Figure 1. Kesterite-type (Hall et al., Reference Hall, Szymanski and Stewart1978) crystal structure (space group
 $I\bar 4$
). In comparison with the cubic sphalerite-type structure, the c-axis is doubled because of cation ordering. Anion coordination tetrahedra are shown for the 2b site only, but are very similar for the other sites. In stoichiometric CZTSSe compounds, 2b is occupied by tin, 2a by copper, 2c also preferably by copper, and 2d preferably by zinc (Schorr, Reference Schorr2011).
$I\bar 4$
). In comparison with the cubic sphalerite-type structure, the c-axis is doubled because of cation ordering. Anion coordination tetrahedra are shown for the 2b site only, but are very similar for the other sites. In stoichiometric CZTSSe compounds, 2b is occupied by tin, 2a by copper, 2c also preferably by copper, and 2d preferably by zinc (Schorr, Reference Schorr2011).
In powder diffraction, the situation is complicated further by the fact that the symmetry of Kesterite is merohedral. As hkl and khl reflections are perfectly superimposed, it becomes impossible for the refinement algorithm to distinguish x from y and 2c from 2d. This also holds in single crystals, as twinning by merohedry is virtually unavoidable in this situation. In a number of cases during the course of preparing this paper, this led to the occupation of 2c and 2d by copper and zinc coming out exchanged, with Zn preferably on 2c and Cu on 2d. Such an apparent exchange of 2c and 2d is structurally equivalent to an exchange of the very barely discriminable anion coordinates x and y.
In such a situation, a single bad dataset or a parameter fixed to an incorrect value can result in refined values, which deviate strongly from the true value, without an obvious reduction in fit quality. It is thus necessary to employ a method of analysis that allows detection of such errors.
B. Linearization
When refining an occupation factor
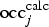 ${\rm occ}_j^{{\rm calc}} $
of a crystallographic site j, the quantity actually refined is the average scattering power of the atoms located at the crystallographic site. For the interaction with X-rays, the scattering power of an atom is described by the atomic form factor f = f
0 + f′ + i·f″. The total scattering power tsp
j
of a crystallographic site is the product of atomic form factor and site occupation factor, occf. In case the site is occupied by multiple chemical species, the total scattering power is the sum of the contribution of all species
${\rm occ}_j^{{\rm calc}} $
of a crystallographic site j, the quantity actually refined is the average scattering power of the atoms located at the crystallographic site. For the interaction with X-rays, the scattering power of an atom is described by the atomic form factor f = f
0 + f′ + i·f″. The total scattering power tsp
j
of a crystallographic site is the product of atomic form factor and site occupation factor, occf. In case the site is occupied by multiple chemical species, the total scattering power is the sum of the contribution of all species
 $${\rm tsp} = \mathop \sum \limits_X {\rm occ}\left( X \right) \cdot f\left( X \right) = \mathop \sum \limits_X {\rm occ}(X) \cdot \left( {\,f_0 \left( X \right) + f^{\prime}\left( X \right) + i \cdot f^{\prime \prime}\left( X \right)} \right).$$
$${\rm tsp} = \mathop \sum \limits_X {\rm occ}\left( X \right) \cdot f\left( X \right) = \mathop \sum \limits_X {\rm occ}(X) \cdot \left( {\,f_0 \left( X \right) + f^{\prime}\left( X \right) + i \cdot f^{\prime \prime}\left( X \right)} \right).$$
If the chemical occupation of the site is unknown, the real total scattering power can be refined in good approximation from a model containing only one element A, which might or might not be a part of the actual mix:
 $${\rm tsp}\left( {{\rm model}} \right) = {\rm occ}^{{\rm calc}} {\rm (}A) \cdot f\left( A \right) = {\rm occ}^{{\rm calc}} (A) \cdot (\,f_0 (A) + f^{\prime}(A) + i \cdot f^{\prime \prime}(A)).$$
$${\rm tsp}\left( {{\rm model}} \right) = {\rm occ}^{{\rm calc}} {\rm (}A) \cdot f\left( A \right) = {\rm occ}^{{\rm calc}} (A) \cdot (\,f_0 (A) + f^{\prime}(A) + i \cdot f^{\prime \prime}(A)).$$
Here and in the following, occcalc is used when referring to site occupation factors of the model, while occ is used for the real site occupation factors in the crystal structure. In the absence of any other sources of error, a refinement will yield
 $${\rm tsp}\left( {{\rm model}} \right) = {\rm tsp,}$$
$${\rm tsp}\left( {{\rm model}} \right) = {\rm tsp,}$$
 $${\rm occ}^{{\rm calc}} (A)\cdot f(A) = \mathop \sum \limits_X {\rm occ}(X) \cdot f(X)\;. $$
$${\rm occ}^{{\rm calc}} (A)\cdot f(A) = \mathop \sum \limits_X {\rm occ}(X) \cdot f(X)\;. $$
If one is interested in the occupation occ of a particular species B, we can split its contribution from the sum at the right-hand side of the equation, as well as the contribution of species A
 $$\eqalign{{\rm occ}^{{\rm calc}} (A) \cdot f\,(A) & = {\rm occ(}B{\rm )} \cdot f\,(B) + {\rm occ}(A)\cdot f\,(A) \cr &\quad + \mathop \sum \limits_{X \ne A,B} {\rm occ}(X) \cdot f\,(X)}$$
$$\eqalign{{\rm occ}^{{\rm calc}} (A) \cdot f\,(A) & = {\rm occ(}B{\rm )} \cdot f\,(B) + {\rm occ}(A)\cdot f\,(A) \cr &\quad + \mathop \sum \limits_{X \ne A,B} {\rm occ}(X) \cdot f\,(X)}$$
that can be rewritten as
 $$\eqalign{{\rm occ}^{{\rm calc}} (A) =& \,{\rm occ(}B{\rm )} \cdot \displaystyle{{\,f\,(B)} \over {\,f\,(A)}} + {\rm occ(}A{\rm )} \cr & + \displaystyle{1 \over {\,f\,(A)}}\mathop \sum \limits_{X \ne A,B} {\rm occ(}X{\rm )} \cdot f\,(X).}$$
$$\eqalign{{\rm occ}^{{\rm calc}} (A) =& \,{\rm occ(}B{\rm )} \cdot \displaystyle{{\,f\,(B)} \over {\,f\,(A)}} + {\rm occ(}A{\rm )} \cr & + \displaystyle{1 \over {\,f\,(A)}}\mathop \sum \limits_{X \ne A,B} {\rm occ(}X{\rm )} \cdot f\,(X).}$$
In conventional X-ray or neutron diffraction, information about the chemical composition of the sample is necessary to determine the element distribution from this relation (Schorr et al., Reference Schorr, Hoebler and Tovar2007; Schorr, Reference Schorr2011). Anomalous X-ray scattering, however, exploits the fact that f′ and f″ are wavelength dependent (Figure 2). It is thus possible to use f(B)/f(A) as a variable against which occcalc(A) can be plotted. A proper definition of this variable is not trivial, as the atomic form factor f = f 0(2θ) + f′(λ) + i·f″(λ) is rather complicated: While the dispersion factor f′ is a constant for a given X-ray wavelength, the base component f 0 is a function of the diffraction angle. Finally, the phase shift described by f″ turns the form factor into a complex function; how this affects the intensities depends on both the site of the atom in the structure and on the Miller indices of the reflection. However, we will show that the simple approximation f ≈ 〈f 0〉 + f′ is sufficient to describe the system, if a properly adjusted value for 〈f 0〉 is chosen. While it is not clear how the average 〈f 0〉 of f 0(2θ) would have to be calculated theoretically, it can easily be derived numerically from the simulated or experimental diffraction patterns of fully ordered structures.

Figure 2. Anomalous X-ray scattering factors f′ and f″ for copper (grey) and zinc (black) in the vicinity of respective K-edge (Cromer and Liberman). Dots indicate energies listed in Table I.
With this approximation, the abovementioned equation gives rise to useful applications. In the simple case of only two elemental species, A and B, it becomes
 $${\rm occ}^{{\rm calc}} \left( A \right) = {\rm occ}\left( B \right) \cdot \displaystyle{{\,f\,(B)} \over {\,f\,(A)}}+ {\rm occ}\left( A \right)\;. $$
$${\rm occ}^{{\rm calc}} \left( A \right) = {\rm occ}\left( B \right) \cdot \displaystyle{{\,f\,(B)} \over {\,f\,(A)}}+ {\rm occ}\left( A \right)\;. $$
Thus, a plot of occcalc(A) over f(B)/f(A) is linear (Figure 3), with occ(B) as slope, occ(A) as zero intercept and the total site occupation occ(A) + occ(B) at f(B)/f(A) = 1. It has to be pointed out that measurements at the absorption edges of both elements A and B can be described with the same line. As f′ close to the edge will become strongly negative, f ≈ 〈f 0〉 + f′ will decrease for this element, while it remains constant in the first approximation for the other element. Thus, the data at the two absorbtion edges will form two arms of the line, with the values at the A-edge at f(B)/f(A) > 1 and those at the B-edge at f(B)/f(A) < 1.

Figure 3. Refined occupation factors for a fully ordered kesterite with exchanged site-occupying species simulated with realistic profile parameters plotted against adjusted scattering factor ratio (full symbols). Ideal limit simulations yield a different slope (open symbols) when plotted against the same ratios. Extreme points deviating from linearity are affected by the f″ jump at the absorption edge; these can be avoided in experiment.
If additional elemental species are present at the particular site, systematic deviations from linearity will occur, expressed by the additional contribution
 $$\displaystyle{1 \over {\,f\,(A)}}\mathop \sum \limits_{X \ne A,B} {\rm occ}\left( X \right) \cdot f\left( X \right).$$
$$\displaystyle{1 \over {\,f\,(A)}}\mathop \sum \limits_{X \ne A,B} {\rm occ}\left( X \right) \cdot f\left( X \right).$$
The effects of this summand on occ(A) are different for the A- and B-arms (Figure 4). In the latter, f(A) is nearly constant. f(B) is not, but does not affect the additional summand. As long as no element X ≠ A, B is present, which happens to have an edge in the observed energy range, all the components of the summand are constant, too. Therefore, the refined occupation value occ(A) will be increased by a constant value for the data points of the B-arm.

Figure 4. (Color online) Refined occupation factors for a simulated fully ordered kesterite. The site indicated in the legend was fully occupied by pure tin (Sn) in the calculation of the pattern, but assumed as Cu or Zn, respectively, in the analysis. Note relatively high deviations from ideal behavior, which is discussed in the text.
In the A-arm the situation is different, as f(A) is reduced close to the absorption edge of element A, while f(B) is fairly constant. As a result the deviation from the ideal line formed by both arms is proportional to the abscissa f(B)/f(A). In addition to the constant increase in the B-arm, the A-arm will show an increased slope; the line will appear bent at f(B)/f(A) = 1.
The amounts of A, B, and other elements X on a particular site can be calculated from the differences between the two arms in a straightforward manner. In the B-arm, the slope is occ(B), while the intercept is occ(A) + occ(X)·f(X)/f(A). In the A-arm, the slope is occ(B) + occ(X)·f(X)/f(B), while the intercept is occ(A). The difference between the two slopes as well as between the two intercepts yields occ(X), provided f(X) is known.
III. ADJUSTMENT OF PARAMETERS
A. Scattering power ratio and slope
The correct numerical value to be used for f 0 can be derived from simulations, in which theoretical diffraction patterns are calculated with one species B occupying a site and subsequently analyzed with another species A in the model. A plot of the results for different values of f′(A) and f′(B) will result in a line with slope and intercept depending on f 0(A) and f 0(B), with slope = 1 and intercept = 0 for the appropriate values. Figure 3 shows this for copper and zinc in kesterites. The structural data of a fully ordered CZTS single crystal (Choubrac et al., Reference Choubrac, Lafond, Guillot-Deudon, Moëlo and Jobic2012) and dispersion correction factors of Cromer and Liberman (Reference Cromer and Liberman1981) were used to calculate powder diffraction data for multiple energies. In this model, the cations were completely ordered, with copper occupying 2a and 2c, and zinc occupying 2d. It is trivial that a Rietveld refinement of the simulated data using the same model yields site occupation factors of 1.0. In subsequent analysis, occupation with copper and zinc was exchanged for a single site in the model, assuming Zn 2a, Zn 2c, or Cu 2d, respectively. This results in refined site occupation factors occcalc different from 1.0. As discussed above, plotting these values against scattering power ratios, f(Zn)/f(Cu) for sites assumed as Cu-occupied in the analysis and f(Cu)/f(Zn) for sites assumed as Zn-occupied, results in linear functions (Figure 3). The appropriate values for 〈f 0〉 are those resulting in a slope = 1.
Ideal powder diffraction data, with full-width at half-maximum (FWHM) → 0 and 2θ-range of 0° through 180°, give
 $$f\,({\rm Cu}) = 20.4 + f^{\prime}\left( {{\rm Cu}} \right)\;{\rm and}\;f\left( {{\rm Zn}} \right) = 21.3 + f^{\prime}\left( {{\rm Zn}} \right).$$
$$f\,({\rm Cu}) = 20.4 + f^{\prime}\left( {{\rm Cu}} \right)\;{\rm and}\;f\left( {{\rm Zn}} \right) = 21.3 + f^{\prime}\left( {{\rm Zn}} \right).$$
There are some changes in refined occcalc with both the high-angle limit and the peak shape and width in the diffraction pattern.
Between the 180° ideal limit and the 105° limit used in our experimental tests, the latter has about 8% lower slope. A realistic peak width and shape increases this effect; typically high-angle peaks are broader and influence the Rietveld fit less than the sharp and intense peaks at low angle. Using the peak shape from our experiments for the simulations, this effect is once again of about 8% strength.
For simulations using the peak width and high-angle limit corresponding to our experimental setup (〈FWHM〉 ≈ 0.1°, 2θ > 105°), the appropriate values were
 $$f\,({\rm Cu}) = 23.2 + f^{\prime}({\rm Cu})\;{\rm and}\;f\,({\rm Zn}) = 24.0 + f^{\prime}\left( {{\rm Zn}} \right).$$
$$f\,({\rm Cu}) = 23.2 + f^{\prime}({\rm Cu})\;{\rm and}\;f\,({\rm Zn}) = 24.0 + f^{\prime}\left( {{\rm Zn}} \right).$$
With these values correct slopes and intercepts were derived from both Cu-modeled sites plotted as function of f (Zn)/f (Cu) and of Zn-modeled sites plotted over f (Cu)/f (Zn).
Both linearity and the values needed for the adjusted scattering factor ratio are mostly independent of the chemical type of the anion in the compound. In particular, in selenides the scattering power of the anion is doubled compared to sulfides. However, the resulting slope of the lines did not change significantly; it was decreased by 0.016(7). The same holds for the intercepts, which increased correspondingly. For any realistic level of experimental accuracy this has to be considered insignificant.
The influence of the f″ component of the scattering factor has been neglected in this analysis. As an imaginary component, the effect of f″ on the scattering power of a particular site is not the simple direct proportionality of the other components. In particular, the effect of f″ on the structure factor depends on the coordinates of the site in the crystal structure. It is thus not possible to give one equation that is valid for all cation sites in the structure. Thus, considering f″ in the analysis leads to a much more complicated recipe. Fortunately, f″ is comparatively small for copper and zinc; its change at the absorption edges is about three electrons. Nonetheless, its effect can be seen (Figure 3): A small discontinuity in the line close to ratio = 1 results from the increased f″(Cu) above the CuKα-edge for all energies at the ZnKα-edge. More pronounced is the offset in the data points at the extremes of both arms, which results from energies just above the absorption edge, where the respective f″ is at a maximum. However, it is trivial to avoid these energies in the experimental data collection. Since above the absorption edge the background is very high because of incoherent scattering, they are inferior in any case. Also the slightly different slopes of the lines for the three different cation sites are a result of the different f″ values of copper and zinc. If these are set to zero in the simulation, the divergence vanishes together with perfect linearity appearing.
As discussed above, occupation of a site by an element different from the two considered in the ratio results in distinct deviations from linearity. The data plotted in Figure 4 result from models of the kesterite structure, in which one additional cation site was assumed to be fully occupied by tin (Sn). The diffraction data simulated from these models were subsequently analyzed in the previously described way, assuming either copper or zinc on the respective site. In the right arms, at the absorption edge of the element assumed in the model, a slope of 1.81(8) is found for assumed Cu, and of 1.82(12) for assumed Zn. Both lines have intercepts not significantly different from zero. In the left arm, the absolute refined occupation factor at the extreme ratios, far away from ratio = 1, is 1.85(2) for Cu and 1.83(3) for Zn. Thus, all parts of the evaluation result in the same value for the occupation factor, which is too high by a factor of 1.83. Accordingly, f(X)/f(Cu) = f(X)/f(Zn) = 1.83 can be used to calculate the site occupation factor of X = Sn as explained in the theory section. This ratio is expected if the scattering factor f(Sn) = f 0(Sn) + f′(Sn) is calculated using f 0(Sn) = 44, which is lower than the number of electrons in the element by approximately the same value of 6 as was used for copper and zinc. Obviously, this cannot be valid with the same constant for all elements; in particular vacancies can be treated as a special case, in which both the number of electrons and the scattering factor are zero.
There are some considerable deviations from the idealized behavior. Most pronounced is the relative reduction in occcalc(Zn) in the data points closest to ratio = 1. The values were derived using energy of 12 398 eV, well above the absorption edges of both copper and zinc. These deviations result from the rather high f″(Sn), not considered in the analysis. This results from the L1-absorption edge of tin at 4465 eV; over the energy range considered in this analysis, f″(Sn) falls from 5.5 to 2.6 with increasing energy. Ironically, it is only above the absorption edges of both copper and zinc that f″ is roughly equal for Sn, Cu, and Zn. These data points thus should be more correct. However, as the general analysis calls for values below the edges, it is prudent to adjust numerical factors accordingly.
B. Experimental values for scattering factors
For all components of the atomic form factor, various tabulated values exist. In this work, we used for the wavelength-independent part f 0 the analytic nine-parameter approximation stored in Fullprof (Rodríguez-Carvajal, Reference Rodríguez-Carvajal2012). The parameters for neutral atoms were used, which were found to work slightly, but not significantly, better than those for cations. The anomalous factors f′ and f″ were taken from the X-ray Anomalous Scattering website of Merritt (Reference Merritt2014). These scattering factor data were calculated using the subroutine library by Brennan and Cowan (Reference Brennan and Cowan1992), in turn using the theoretical approximation developed by Cromer and Liberman (Reference Cromer and Liberman1981). Values between tabulated energies were derived by linear interpolation.
Cromer and Liberman theory gives accurate values far from an absorption edge but does not account for the effects of neighboring atoms, which can be very substantial near an absorption edge. For the purpose of the analysis detailed in here, this effect is best gauged from a site, which is fully occupied only by the one chemical species whose form factor parameters are tested. In this case and in the absence of any other sources of error, the occupation factors refined using the scattering factor for the correct element should all give the correct occcalc = occ = 1. The refined value might deviate from 1 if either the site occupation factor is <1 or if correlated parameters, e.g. the thermal displacement parameters, are incorrect. But even in these cases the refined values should be constant. The crystal structure of the compound used for this calibration has to be similar to the structure of the real samples for values to be transferable.
A sufficient number of different energies both close to the edge and far away from it should be measured to allow distinction between different sources of error: (1) Estimated standard deviations from Rietveld refinements tend to be too small (Bérar and Lelann, Reference Bérar and Lelann1991); the real uncertainty of the results is higher. For those values far from the absorption edge, this is the factor determining the data scattering. (2) If the anomalous scattering factors close to the absorption edge are significantly different from theory, then the refined occ at these energies deviate from the constant more than average. As the deviation from theory is probably systematic, the horizontal line can be expected to gain a hockey stick bend. (3) The relative position of the absorption edge might be shifted. This can happen from a real shift of the absorption edge compared with the values for the pure element (Kumar et al., Reference Kumar, Nagarajan and Sarangi2013). It might also be the result of an error in the energy calibration of the experiment. (4) If the assumption of a fully ordered structure is not correct and some amount of other chemical elements occupies the site, the deviations from linearity described in the previous section will be observed.
It should be obvious that the high number of potential systematic errors makes it necessary to measure a rather large number of different energies both close to the edge and farther away from it.
For the examination of the f′-values of copper a stoichiometric CZTS sample cooled down at a rate of 1 K h−1 was used. Neutron diffraction analysis (Schorr et al., Reference Schorr, Hoebler and Tovar2007) showed that in this sample the 2a position is fully occupied by copper only. All occcalc data points far away from the CuK edge are constant within ±0.01 (Figure 5). The average occupation calculated from these values is slightly increased, with 〈occcalc〉 = 1.021(7). As discussed above, this might have any number of reasons and it is not justified to use f′ to adjust this. In contrast, the two refined occupancies close to the CuK edge are significantly increased. This indicates too low scattering power attributed to copper in the model, thus f′ too negative. To bring these values down to the average, the values of f′ had to be changed, at 8969 eV from −6.5604 to −6.26, at 8974 eV from −7.4207 to −6.70 (Table I). Experimental f′ at the CuK edge is less pronounced than predicted by the theory. This is expected, as the real structure effects will tend to broaden the sharp theoretical curve.

Figure 5. Experimental refined occupation factors of atomic site fully occupied by either copper or zinc only. For energies close to the absorption edge, f′ had to be adjusted, so that always the same occupation factor results. This is independent of systematic errors that can affect the absolute value of the total occupation factor.
Table I. Anomalous scattering factors f′ of copper and zinc (Cromer and Liberman).

Values marked * have been adjusted from the original value given in brackets.
For the scattering factors of Zn a sample of pure sphalerite, ZnS, was analyzed. Sphalerite has strong cleavability along {110} (Kleeberg and Mibus, Reference Kleeberg and Mibus2010), which was considered in the Rietveld analysis by a preferred orientation model using the modified March function. The resulting occupation factors of zinc are slightly below 1, with 〈occcalc〉 = 0.993(9). Significant deviation from the theoretical value of f′ was observed at the energy closest to the edge; f′ at 9654 eV was changed from −7.5581 to −7.20.
IV. EXAMPLES OF PRACTICAL APPLICATION
The method described was applied to a well-characterized CZTSSe sample, a monograin powder previously analyzed by neutron powder diffraction. Cation ratios Cu/(Zn + Sn) = 0.944 and Zn/Sn = 1.039 of the kesterite-type phase were determined by wavelength dispersive X-ray spectroscopy (WDX). This corresponds to an A-type kesterite (Lafond et al., Reference Lafond, Choubrac, Guillot-Deudon, Deniard and Jobic2012) and a chemical composition as Cu1.942Zn1.049Sn1.009S3.288Se0.712. The sample contains small amounts of two secondary phases, determined by WDX and PXRD, 5.1(1.8)% ZnS-type phase Zn0.95Cu0.04Sn0.01S0.9Se0.1 and 2.8(5)% Cu2SnS3-type phase Cu1.5Zn0.5SnS2.3Se0.7. Visible peaks of the latter were excluded from the refinement. The peaks of the ZnS-type phase overlap with the main peaks of the kesterite phase. Considering this phase resulted in negligible changes in the results. The procedure applied in this particular case is intended as an example only; it should be adapted to the particular experiment:
-
(1) Data collection: Anomalous X-ray powder diffraction data (AXPD) were collected at the diffraction end station of the KMC-2 beamline (Erko et al., Reference Erko, Packe, Hellwig, Fieber-Erdmann, Pawlizki, Veldkamp and Brennan2000; Helmholtz-Zentrum Berlin für Materialien und Energie, 2016) at BESSY II (Berlin, Germany). KMC-2 provides X-ray radiation with both very stable energies and intensities. The accessible energy range of 4–14 keV is ideally suited for the K-edges of Cu (8979 eV) and Zn (9659 eV). In the experiments described here eight different energies were used, one far below the absorption edge (8048 eV), three each at 60, 10, and 5 eV below the absorption edges of copper and zinc, and one above (12 398 eV). Total data collection time for one sample was 5 h, for a 2θ-range of 5–104° and about 60 000 counts in the strongest peak at an integration step size of 0.01°. The sample was in symmetric reflection geometry at ambient condition. Sufficient particle statistic was ensured by rotating the sample around an excentric point outside of the illuminated area, and by using a high number of θ–2θ-positions in combination with a Vantec 2000 area detector with 12° acceptance angle both vertically and horizontally. As in this geometry the symmetric reflection condition is not strictly conserved, an appropriate absorption correction was applied during data refinement (Wronski et al., Reference Wronski, Wierzbanowski, Baczmanski, Lodini, Braham and Seiler2009).
-
(2) LeBail refinements were done using Fullprof.2k (Rodríguez-Carvajal, Reference Rodríguez-Carvajal2001) to determine the optimum fit profile model. In this case, a Pearson VII peak shape was adopted. Systematic peak shifts and anisotropic peak broadening were tested, but found to be insignificant. Background was interpolated between positions with no or little intensity contribution from peaks. Regions containing small peaks from the impurity were excluded from the refinement. Peak width, asymmetry, and background were refined and fixed for subsequent Rietveld analysis.
-
(3) The structure model was taken from the ordered CZTS kesterite ICSD 262388 (Choubrac et al., Reference Choubrac, Lafond, Guillot-Deudon, Moëlo and Jobic2012), which was refined from single-crystal data. The anisotropic displacement parameters of all atoms were retained. The anion occupancy was modified to reflect the composition of the sample; differences resulting from nominal (S = 3.25, Se = 0.75) or microprobe (S = 3.288, Se = 0.712) composition were found insignificant. Cation occupation of the sites were assigned as Cu 2a, Cu 2c, Zn 2d, Sn 2b, each fully occupied, that is the ideal ordered kesterite-type structure. In this stage, only the 8048 eV dataset was used, where the unknown Cu/Zn distribution has little influence. The dataset at 12 398 eV gave results with higher uncertainties because of the increased background from copper and zinc fluorescence. The use of an energy-discriminating detector might be useful here. Scale factor and lattice were set as refinable parameters. Structural parameters refined in the preparatory stage were S, Se coordinates xyz and an overall displacement parameter. The occupancy of Sn 2b was tested, but did not differ significantly from full occupation. The same holds for preferred orientation. The refined parameters were subsequently transferred to all datasets and fixed for subsequent analysis.
-
(4) Occupation factors of Cu 2a, Cu 2c, Zn 2d were independently refined for all eight datasets.
-
(5) Subsequent data analysis was done using Origin 9.0. The results from step 4 were plotted (Figure 6) as function of f(Zn)/f(Cu) or f(Cu)/f(Zn), using the scattering factors f(Cu) = 23.2 + f′(Cu) and f(Zn) = 24.0 + f′(Zn), as determined from simulations. The data were checked for linearity within statistical uncertainty and for impossible trends, in particular negative slopes, whose presence would have indicated problems in either experiment or analysis procedure. Regression lines for the three sites were fitted by least-squares procedure, with the data points weighted by their Rietveld-derived e.s.d.s.
-
(6) The results are given in Table II, together with occupancies derived from a previous analysis by neutron diffraction. It might be necessary to normalize the results to comply with total composition from chemical analysis or charge balance. This was not necessary in this case. In an A-type kesterite, expected defects because of deviation from stoichiometry are copper vacancies (V Cu) and ZnCu, where extra zinc is located at Cu-sites. Both are generally concentrated at the 2a site (Lafond et al., Reference Lafond, Choubrac, Guillot-Deudon, Deniard and Jobic2012). The latter are confirmed in this AXRPD analysis. The analysis gives no indication of vacancies; however, their expected number is below uncertainties. Additional Cu/Zn disorder on the 2c and 2d sites is found correctly.

Figure 6. Results of an exemplary analysis with fitted linear dependency (bold solid line). Shown for clarity are the expected line from neutron analysis (thin solid line) and the lines with slope 0 and 1 representing full occupation with Cu or Zn (dotted lines).
Table II. Results of AXRPD analysis and neutron analysis.

Note that neutron results given for comparison are necessarily normalized to the results of the chemical analysis. The 2b site is occupied by tin.
V. CONCLUSION
Anomalous diffraction at the absorption edges of copper and zinc allows quantitative determination of the cation disorder in CZTSSe materials. The attainable accuracy is at the same level as can be reached by neutron diffraction. Owing to the particular crystal structure of kesterites, strong correlations between structural parameters are unavoidable. Extremely careful experimentation and analysis is needed to avoid and detect systematic errors. Even under the best conditions, some uncertainty about the accuracy of the obtained absolute values of site occupation factors remains. Combining the raw results with additional information, e.g. chemical analysis, should allow resolving this. The same holds for parametric studies, in which multiple samples are measured and analyzed in the same way.
ACKNOWLEDGEMENTS
Measurements were carried out at the Diffraction instrument at KMC-2 beamline at Helmholtz-Zentrum Berlin für Materialien und Energie. We thank HZB for the allocation of synchrotron radiation beamtime.








