I. INTRODUCTION
Metal–organic frameworks (MOFs) and zeolites have been thoroughly studied for a wide variety of applications including catalysis (Chen et al., Reference Chen, Zhangm, Jiao and Jiang2018), gas storage (Mason et al., Reference Mason, Veenstra and Long2014), gas separation (Lorzing et al., Reference Lorzing, Trump, Brown and Bloch2017), and sensing (Davis, Reference Davis2002). The properties of these materials vary significantly based on the metal cations that comprise them. In the case of hybrid metal/organic materials, such as three-dimensional (3D) MOFs, two-dimensional (2D) MOFs, and discrete porous coordination cages, properties can further be tuned via the organic bridging ligands. All three classes of these hybrid materials are typically synthesized via solvothermal methods and display Langmuir surface areas greater than 2500 m2 g−1. However, these solvothermal syntheses typically require high boiling point, highly-polar organic solvents at temperatures up to 300 °C. In order to realize the large-scale synthesis of metal–organic materials, alternative greener methods to circumvent these conditions have been attempted. Mechanochemistry has shown utility in this regard. Here, solvent-free or nearly solvent-free synthetic conditions are employed to afford material in quantitative yield with significantly shorter reaction times (Son et al., Reference Son, Kim, Kim and Ahn2008; Friscic, Reference Friscic2010; Crawford et al., Reference Crawford, Casaban, Haydon, Giri, Mcnally and James2015; Kradeniz et al., Reference Kradeniz, Howarth, Stolar, Islamoglu, Dejanovic, Tireli, Wasson, Moon, Farha, Friscic and Uzarevic2018; Prochowicz et al., Reference Prochowicz, Nawrocki, Terlecki, Marynowski and Lewinski2018). Mechanochemistry, which can entail grinding, extrusion, and ball-milling, has been applied to a wide variety of porous materials from 3D- to zero-dimensional metal–organic materials, porous-organic cages, and molecular cocrystals. In all cases, milled products produce very crystalline X-ray diffraction patterns that can be analyzed and compared to the simulated diffraction pattern (Carrington et al., Reference Carrington, Vitorica-Yrezabal and Brammer2014). We have previously shown that phase control of lower dimensional metal–organic materials can be achieved by functionalizing 5-hydroxyisophthalic acid with linear alkoxide groups (Barreda et al., Reference Barreda, Bannwart, Yap and Bloch2018a, Reference Barreda, Taggart, Rowland, Yap and Blochb). In this study we show how increasing the length of the alkoxide chain and introducing steric bulk prevents the formation of 2D tetragonal sheets and rather forms a more stable 2D hexagonal material upon heating. Under solvent-free conditions, bulky tert-butylamide- and pentoxide-functionalized isophthalic acid ligands afforded discrete porous cages and hexagonal 2D materials, respectively (Barreda et al., Reference Barreda, Bannwart, Yap and Bloch2018a, Reference Barreda, Taggart, Rowland, Yap and Blochb). In order to determine if tetragonal 2D MOFs are isolable via mechanochemical reaction of copper(II) with short-chain alkoxide groups, we followed the ball-mill reaction of these via powder X-ray diffraction (PXRD) methods (Figure 1).
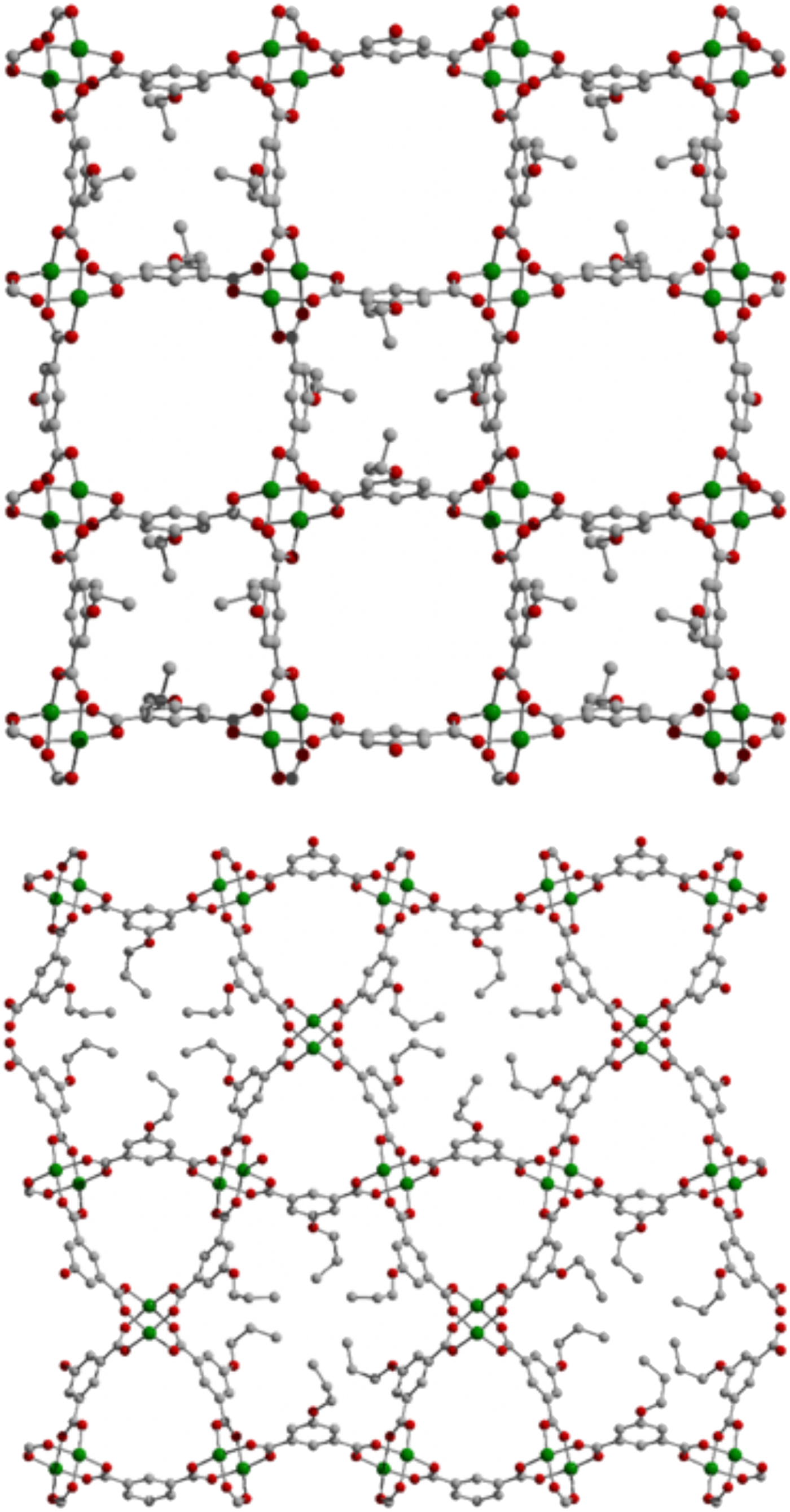
Figure 1. Tetragonal (top) and hexagonal (bottom) 2D MOFs based on functionalized isophthalic acid ligands and copper(II) paddlewheels; copper metals are represented by green spheres, carbon by gray spheres, and oxygen by red spheres. Under solvothermal reaction conditions, the hexagonal material is the favored phase.
II. EXPERIMENTAL
A. Synthesis of 5-ethoxy-1,3-benzenedicarboxylic acid
Dimethyl 5-hydroxyisophthalate (5.10 g, 24.3 mmol), potassium carbonate (5.20 g, 37.6 mmol), and ethyl iodide (2.2 ml, 27.4 mmol) were added to a 250 ml round-bottom flask with 100 ml of acetone and stirred at 40 °C for 12 h. A colorless precipitate was separated via vacuum filtration. Acetone and excess ethyl iodide were pumped off with reduced pressures at 60 °C. Potassium hydroxide (0.617 g, 11.0 mmol) in 20 ml of H2O was added to the filtrate with another 30 ml of acetone and stirred for 12 h at 50 °C. The solution was concentrated to remove the acetone, and then acidified to a pH of 1. The tan precipitate was isolated via vacuum filtration. Precipitate was thoroughly washed with CHCl3 and dried to yield 87% of product. 1H-NMR (400 MHz, DMSO-d6): δ = 13.30 (s, 2H, OH), 8.05 (s, 1H, CH arom), 7.61 (s, 2H, CH arom), 4.05 (q, 2H, CH2), 1.72 (sextet, 2H, CH2), 1.37 (sextet, 2H, CH2), 1.36 (sextet, 2H, CH2), 0.88 (t, 3H, CH3).
B. Synthesis of 5-propoxy-1,3-benzenedicarboxylic acid
Dimethyl 5-hydroxyisophthalate (5.98 g, 28.5 mmol), potassium carbonate (5.6 g, 40.6 mmol), and propyl iodide (3.1 ml, 37.7 mmol) were added to a 250 ml round-bottom flask with 100 ml of acetone and stirred at 40 °C for 12 h. A colorless precipitate was separated via vacuum filtration. Potassium hydroxide (0.617 g, 11.0 mmol) in 20 ml of H2O was added to the filtrate and stirred for 12 h at 50 °C. The solution was concentrated to remove the acetone, and then acidified to a pH of 1. The tan precipitate was isolated via vacuum filtration. Precipitate was thoroughly washed with CHCl3 and dried to yield 68% of product. 1H-NMR (400 MHz, DMSO-d6): δ = 13.30 (s, 2H, OH), 8.05 (s, 1H, CH arom), 7.61 (s, 2H, CH arom), 4.05 (q, 2H, CH2), 1.72 (sextet, 2H, CH2), 1.37 (sextet, 2H, CH2), 1.36 (sextet, 2H, CH2), 0.88 (t, 3H, CH3).
C. Synthesis of 5-butoxy-1,3-benzenedicarboxylic acid
Dimethyl 5-hydroxyisophthalate (5.32 g, 25.3 mmol), potassium carbonate (5.51 g, 39.9 mmol), and butyl iodide (3.7 ml, 28.3 mmol) were added to a 250 ml round-bottom flask with 100 ml of acetone and stirred at 40 °C for 12 h. A colorless precipitate was separated via vacuum filtration. Potassium hydroxide (0.617 g, 11.0 mmol) in 20 ml of H2O was added to the filtrate and stirred for 12 h at 50 °C. The solution was concentrated to remove the acetone, and then acidified to a pH of 1. The tan precipitate was isolated via vacuum filtration. Precipitate was thoroughly washed with CHCl3 and dried to yield 75% of product. 1H-NMR (400 MHz, DMSO-d6): δ = 13.30 (s, 2H, OH), 8.05 (s, 1H, CH arom), 7.61 (s, 2H, CH arom), 4.05 (q, 2H, CH2), 1.72 (sextet, 2H, CH2), 1.37 (sextet, 2H, CH2), 1.36 (sextet, 2H, CH2), 0.88 (t, 3H, CH3).
D. Synthesis of 5-pentoxy-1,3-benzenedicarboxylic acid
Dimethyl 5-hydroxyisophthalate (5.423 g, 25.8 mmol), potassium carbonate (4.3 g, 31.2 mmol), and R iodide (3.7 ml, 28.3 mmol) (R = ethyl, propyl, butyl, pentyl) were added to a 250 ml round-bottom flask with 100 ml of acetone and stirred at 40 °C for 12 h. A colorless precipitate was separated via vacuum filtration. Potassium hydroxide (0.617 g, 11.0 mmol) in 20 ml of H2O was added to the filtrate and stirred for 12 h at 50 °C. The solution was concentrated to remove the acetone, and then acidified to a pH of 1. The tan precipitate was isolated via vacuum filtration. Precipitate was thoroughly washed with CHCl3 and dried to yield 85% of product. 1H-NMR (400 MHz, DMSO-d-6): δ = 13.30 (s, 2H, OH), 8.05 (s, 1H, CH arom), 7.61 (s, 2H, CH arom), 4.05 (q, 2H, CH2), 1.72 (sextet, 2H, CH2), 1.37 (sextet, 2H, CH2), 1.36 (sextet, 2H, CH2), 0.88 (t, 3H, CH3).
E. Synthesis of 5-OEt 2D sheets
Cu(OAc)2·H2O (0.430 g, 2.15 mmol) and 5-ethoxy-1,3-benzenedicarboxylic acid (5-OEt-1,3-H2bdc) (0.460 g, 2.19 mmol) were added to a 65 ml steel vial with two 1 cm diameter steel ball bearings. The resulting mixture was milled for 60 min at 12-min intervals and characterized by PXRD once before starting the milling process. After 12 min, a powder aliquot was taken from the steel container and characterized by PXRD.
F. Synthesis of 5-OPr 2D material
Cu(OAc)2·H2O (0.428 g, 2.14 mmol) and 5-propoxy-1,3-benzenedicarboxylic acid (5-OEt-1,3-H2bdc) (0.497 g, 2.22 mmol) were added to a 65 ml steel vial with two 1 cm diameter steel ball bearings. The resulting mixture was milled for 60 min at 12-min intervals and characterized by PXRD once before starting the milling process. After 12 min, a powder aliquot was taken from the steel container and characterized by PXRD.
G. Synthesis of 5-OBut 2D sheets
Cu(OAc)2·H2O (0.429 g, 2.15 mmol) and 5-butoxy-1,3-benzenedicarboxylic acid (5-OEt-1,3-H2bdc) (0.515 g, 2.16 mmol) were added to a 65 ml steel vial with two 1 cm diameter steel ball bearings. The resulting mixture was milled for 60 min at 12-min intervals and characterized by PXRD once before starting the milling process. After 12 min, a powder aliquot was taken from the steel container and characterized by PXRD.
H. Synthesis of 5-OPent 2D sheets
Cu(OAc)2·H2O (0.420 g, 2.10 mmol) and 5-pentoxy-1,3-benzenedicarboxylic acid (5-OEt-1,3-H2bdc) (0.540 g, 2.12 mmol) were added to a 65 ml steel vial with two 1 cm diameter steel ball bearings. The resulting mixture was milled for 60 min at 12-min intervals and characterized by PXRD once before starting the milling process. After 12 min, a powder aliquot was taken from the steel container and characterized by PXRD.
I. Ball-milling
Mechanochemical reactions were performed in a 65 ml stainless steel vessel containing two 1-cm stainless steel ball bearings and milled in a SPEX Sample Prep at 12 min intervals up to a total reaction time of 60 min.
J. PXRD measurements
PXRD experiments were carried out at room temperature using a Rigaku Miniflex powder diffractometer using filtered Cu-Kα radiation (λ = 1.54056 Å). The data were collected from 2θ ranging from 5 to 30° with a step size of 0.05 and 0.1 s per step counting time. The voltage was set at 30 kV and current at 15 mA.
III. RESULTS AND DISCUSSION
In order to accurately determine reaction times, it was necessary to set up an experiment that would allow for the isolation of aliquots at 12-min time intervals. An attempt to keep the weight of the metal salt as consistent as possible was made between 420 and 430 mg with 1 M equivalent for each ligand. Upon adding starting materials to the steel container, they were briefly stirred with a spatula prior to ball-milling for the t = 0 time point. Once the aliquot was taken out, the sample was capped and the rest of the sample was ball-milled for 12 more minutes and another aliquot was taken out and measured using the diffractometer. The same procedure was followed until 60 min of run time had been acquired.
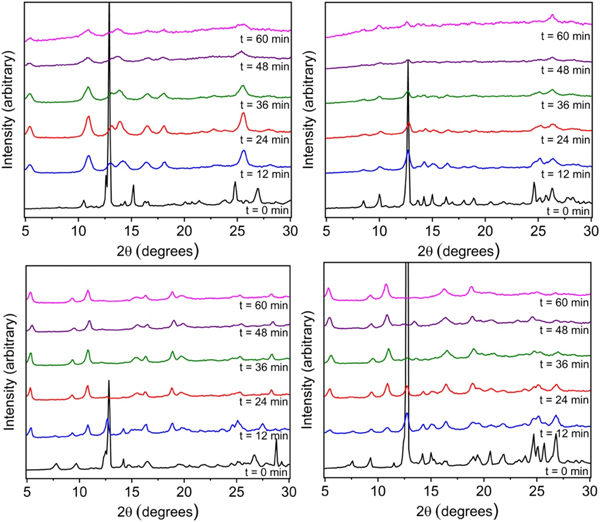
As shown in Figure 2 for the ethoxy-functionalized isophthalic acid, the peaks associated with copper acetate and organic ligand are largely absent after just 12 min of milling time with concomitant appearance of a number of additional peaks. Specifically, the peak at 10.2° shifted to 11° and the copper acetate peak at 12.7° decreased significantly. In addition, the appearance of a broad peak at 5.2° was observed. This peak became more intense with additional milling. Peaks at 14°, 18°, and 23°, which correspond to the hexagonal MOF, reached maximum intensity at 24 min of milling. After 24 min, the peak intensity of the final product diminished. The final product's powder pattern is in good agreement with that of previously reported copper and 5-ethoxyisophthalic acid 2D hexagonal sheets (Figure 3).
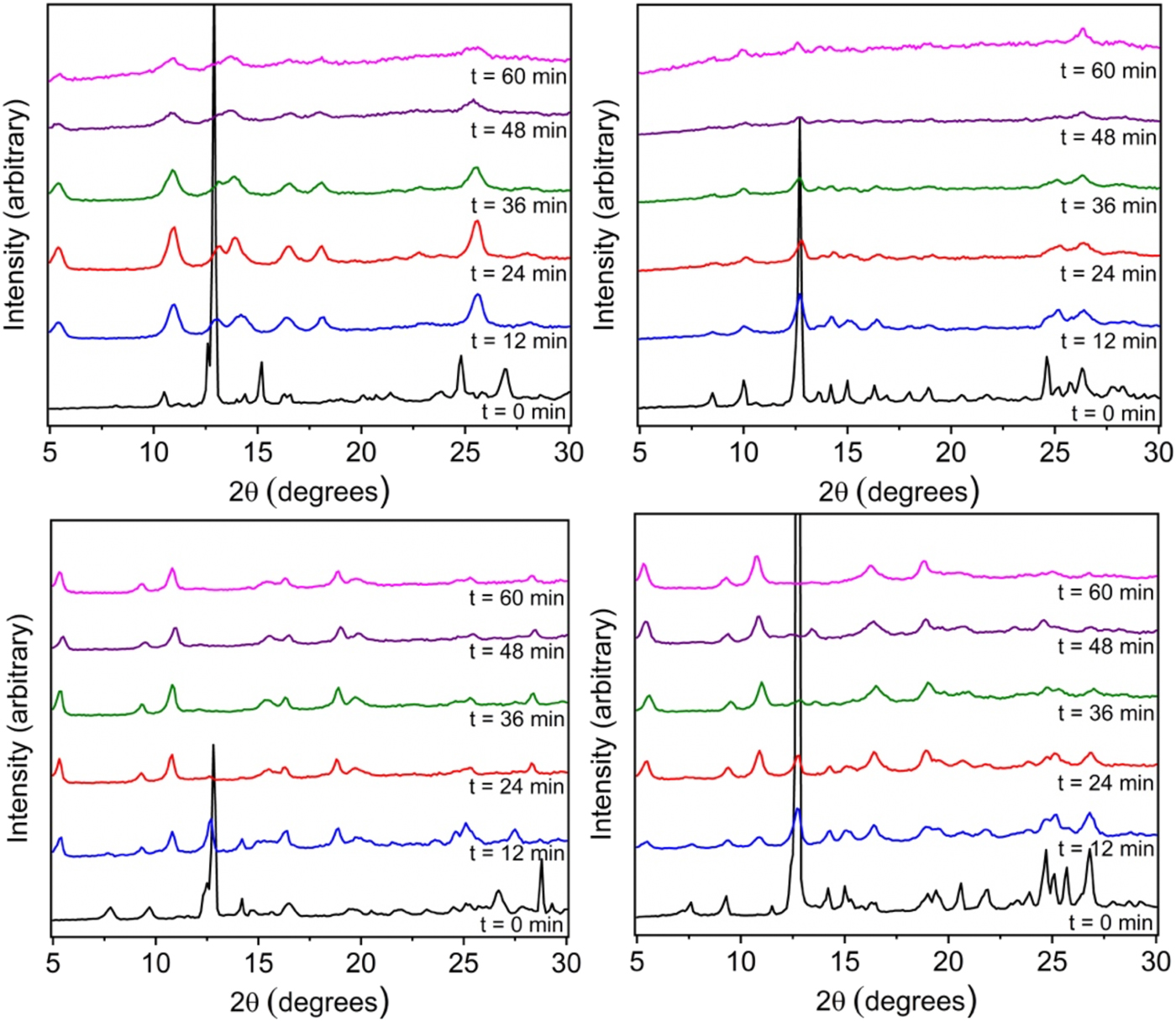
Figure 2. PXRD patterns of materials ball-milled at the indicated times. The plots correspond to ethoxy- (top left), propoxy- (top right), butoxy- (bottom left), and pentoxy- (bottom right) functionalized isophthalic acid.

Figure 3. PXRD patterns for ethoxy- (left), butoxy- (middle), and pentoxy- (right) based 2D MOFs. The ticks indicate symmetry-allowed reflections while the asterisk indicates an impurity.
A similar experimental procedure was followed for propoxy, butoxy, and pentoxy based materials. In the case of propoxy, there was no apparent change in the powder pattern after milling for 60 min. The intensity of the copper acetate and ligands decreases and no new peaks are shown in the powder pattern. However, for the butoxy based ligand, we see a clear transformation of starting materials to product within 12 min of milling. For butoxy, 36 min of milling was enough to complete the reaction. In fact, further milling did not decrease the crystallinity of the material showing greater stability to milling as compared to the ethoxy ball-milled material. Starting material peaks for the butoxy ligand and copper acetate showed significant intensity at 0 min and after 12 min of milling the peaks at 6.7° and 9.8° shifted to 9.5° and 10.8°, respectively, while the peaks at 5.2° and 18.2° appeared. At 36 min of ball-milling the copper acetate peak at 12.7° was no longer present in the powder pattern. The powder pattern at 36 min matched that of previously reported copper and 5-butoxyisophthalic acid 2D hexagonal sheets.
The experiment yielded the same 2D hexagonal sheet when pentoxy isophthalic acid and copper acetate were ball-milled. The powder pattern shows peaks at 7.5° and 9.2° which shift after 12 min of milling to 9.5° and 11°, respectively. The typical peak at 5.2° was present at 12 min and reached maximum intensity at 48 min of ball-milling. In fact, the reaction was fully complete at 60 min of milling when the copper acetate peak disappeared completely. Crystallinity of the final product seemed to remain intact after 60 min showing similar stability characteristics to the butoxy material.
When comparing the PXRD patterns of the ball-milled materials to the simulated PXRD pattern of the alkoxy 2D hexagonal sheets, there is largely excellent agreement. This is particularly the case for the butoxy- and pentoxy-based solids. However, in comparing the PXRD pattern of the ethoxide-based MOF, there are a number of diffraction peaks that cannot be attributed to the hexagonal phase. This is not unexpected, as the limited ligand bulk of the ethoxide group is not competent for phase discrimination. This is likely similarly the case for the propoxide-based solid as the PXRD pattern suggests limited crystallinity even after 60 min of milling.
IV. CONCLUSION
The foregoing results demonstrate the utility of mechanochemical methods for the synthesis of 2D MOFs based on copper paddlewheel units and functionalized isophthalic acid ligands. Although solvothermal routes afford tetragonal sheets, hexagonal sheets, or discrete porous coordination cages, depending on reaction temperature and solvent conditions, ball-milling ethoxide through pentoxide ligands favors the formation of 2D hexagonal frameworks. It is expected that these materials, which are widely applicable for a variety of separation applications, will be compatible with longer linear functionalization. Importantly, this work illustrates the power of solvent-free syntheses for potential large-scale MOF production.
SUPPLEMENTARY MATERIAL
The supplementary material for this article can be found at https://doi.org/10.1017/S0885715619000228
ACKNOWLEDGEMENTS
We are grateful to the University of Delaware for generous start-up funds that made this work possible. A portion of this work was supported by the National Institutes of Health under award number P20GM104316.