



I. INTRODUCTION
The crystal structure of Ag3O was determined by Beesk et al. (Reference Beesk, Jones, Rumpel, Schwarzmann and Sheldrick1981) using XRD on a crystal grown by the hydrothermal reaction of AgO on a Ag substrate at 4 kbar for several days.Footnote 1 No other published study has identified Ag3O prior to the present study. The structure consists of a sublattice of hexagonal closest-packed (hcp) Ag with a two-layer repeat and oxygen occupying two-thirds of the octahedral interstices in every other adjacent closest-packed layer [Figures 1(a) and 1(b)].

Figure 1. (Color online) The structure of Ag3O determined by Beesk et al. (Reference Beesk, Jones, Rumpel, Schwarzmann and Sheldrick1981); light (grey) spheres are hcp Ag and dark (red) spheres are oxygen; the a axis is red, the b axis is green, and the c axis is blue. (a) Perspective view and (b) c axis projection showing the tunnels formed by the empty octahedral interstices sharing rotated trigonal faces (blue).
Prior to the Ag3O structure determination, X-ray powder diffraction (XRPD) patterns of silver oxide powders produced at 115–125 kbar and 1100–1400 °C were reported as “Ag2O II” and “high-pressure modification” by Kabalikina et al. (Reference Kabalikina, Popova, Serebryanaya and Vereshchagin1963). The Ag2O II crystal structure was described as hcp Ag with oxygen occupying all octahedral interstices in every other adjacent, cp layer; this layered structure is isostructural with Ag2F, an inverse-CdI2 structure type. Similar powder patterns were reported by Birkenberg and Schwarzmann (Reference Birkenberg and Schwarzmann1974) for “hexagonal Ag2O” synthesized by hydrothermal reactions of AgO and Ag2O on Ag substrates for 6 weeks at 3.7 kbar and temperature gradients from 79 to 52 °C.
Beesk et al. (Reference Beesk, Jones, Rumpel, Schwarzmann and Sheldrick1981) found the hexagonal unit cell reported for Ag2O II at ambient conditions was a subcell of their Ag3O P  $\bar{3}$1 m cell with a = 5.318(2) Å and c = 4.951(2) Å where the a axis is
$\bar{3}$1 m cell with a = 5.318(2) Å and c = 4.951(2) Å where the a axis is 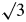 $\surd 3\;$times the a axis of the subcell and is rotated by 60° around the subcell c axis. Weak peaks generated by the ordered occupation of oxygen in two-thirds of the octahedral interstices in Ag3O were not observed in the Debye–Scherrer patterns of the material synthesized in the high-pressure and hydrothermal experiments. However, the very low signal-to-background in the published Debye–Scherrer images would have obscured these weak peaks if present.
$\surd 3\;$times the a axis of the subcell and is rotated by 60° around the subcell c axis. Weak peaks generated by the ordered occupation of oxygen in two-thirds of the octahedral interstices in Ag3O were not observed in the Debye–Scherrer patterns of the material synthesized in the high-pressure and hydrothermal experiments. However, the very low signal-to-background in the published Debye–Scherrer images would have obscured these weak peaks if present.
In the present study, a suite of Ag3O-rich powders was synthesized by heating jet-milled, magnetron-sputtered films from room temperature to 148 °C at atmospheric pressure in an atmosphere composed of either ambient air or N2 for time periods from hours to several years. The reactant jet-milled films were composed of a majority of Ag2O with the cuprite structure and ccp Ag, as well as a minor amount of Ag3O. Ag3O was also produced from the jet-milled powders by ball milling and by sonication. The phase composition and unit-cell parameters of these products were measured with in situ and ex situ experiments from −186 to 293 °C as a function of temperature and time. The thermal reaction was studied as a function of time at 100 and 130 °C.
II. EXPERIMENTAL
A. Physical vapor deposition and jet-milling of the reactant thin film
Ag3O powders were synthesized by heating jet-milled films composed of a mixture of Ag2O and ccp Ag in air and nitrogen. The films were provided by NUCRYST Pharmaceuticals Inc. and were grown by reactive, direct-current, magnetron sputtering of silver (99.99 wt%) onto stainless steel. The atmosphere was composed of approximately 2 vol% oxygen and 98 vol% argon, with a nominal pressure of 5.3 × 10−5 atm and gas-flow rates for Ar and O2 of 7400 and 160 standard cm3 min−1, respectively. The temperature of the stainless-steel substrate was 35 ± 3 °C. The films were removed from the stainless-steel substrate by scraping with a stainless-steel blade.
The removed films were jet milled using a Micron-Master 8” orbital jet mill by Jet Pulverizer Company. Compressed ambient air was used for jet milling. Jet-milled films were used as reactants (Table I) to produce the samples described in Table II. U1 and U2 were products from reactant thin films similar to those included in Table I; however, the phase composition of these films was unavailable.
Table I. Estimated phase compositions of jet-milled films.

Table II. Ex situ reaction-product identifications, reaction times, temperatures and atmospheres, and estimated phase compositions of thermally reacted, sonicated, and ball-milled reactant films.

B. Diffractometer configuration and in situ temperature correction
XRPD patterns were collected using transmission and Bragg–Brentano configurations of Panalytical X'Pert Pro MPD and Empyrean diffractometers. CuKα X-rays generated at 45 kV and 40 mA were focused on an X'Celerator detector using an elliptically graded W/Si multilayer mirror for the transmission configuration. Transmission specimens were packed between thin polymer films. The Bragg–Brentano configuration used a Ni filter, an X'Celerator, fixed slits (0.125° divergence, 0.25° incident-beam antiscatter, and 5.0 mm diffracted-beam antiscatter), and specimens packed in a 0.2-mm well in a silicon zero-background holder.
In situ heating and cooling experiments were completed with Anton Paar TTK 450 and THC stages in Bragg–Brentano configuration. The temperature of the specimen was measured using a 100 Ω platinum resistive-temperature probe (Pt-100) positioned in a nickel-coated copper holder under the specimen well. A thin layer of heat-conducting grease was applied to the bottom of the holder. Humidity for in situ experiments was measured with an HC2-C05 capacitive humidity probe housing a Pt-100 sensor in the THC chamber. Data was displayed by a Rotronic HF5 transmitter. Humidity was generated by a VTI RH-200.
Temperatures above ambient were corrected using in situ XRPD of several organic melting-point standards and the thermal expansion of Ag metal determined by Suh et al. (Reference Suh, Ohta and Waseda1988) from 25 to 1228 °C using XRPD (Supplementary Figure S1). To account for the current CuKα 1 used for whole-pattern refinements (section II.C), the thermal-expansion reference value for Ag at 25 °C was the average Ag unit-cell length 4.0865404 Å from Yoder-Short (Reference Yoder-Short1993) rescaled to 4.086527 Å by a multiplication factor of 0.9999966 = 1.5405929 (current CuKα 1) ÷ 1.5405981 (Yoder-Short, Reference Yoder-Short1993).
Temperatures below room temperature were calibrated using the reversible and sharp transformation of ammonium sulfate from space group Pnam at room temperature to Pna21 at −50.0 °C (Supplementary Figure S2; Schlemper and Hamilton Reference Schlemper and Hamilton1966; Kosova et al., Reference Kosova, Emelina and Bykov2014). Low temperatures were also calibrated using adamantine which transforms from ccp at room temperature to tetragonal at −65.2 °C (Supplementary Figures S3 and S4; Nordman and Schmitkons, Reference Nordman and Schmitkons1965; van Ekeren et al., Reference van Ekeren, van Genderen and van den Berg2006). The maximum correction of 3 °C for the lowest temperature observed in the present study, 189 °C, was extrapolated from a quadratic regression (Supplementary Figure S1).
C. Whole-pattern refinement
XRPD patterns were processed using TOPAS V4.2 (Kern et al., Reference Kern, Coelho, Cheary, Mittemeijer and Scardi2004; Bruker-AXS, 2009). Unit-cell parameters were determined using converged Pawley (Reference Pawley1981) refinements. The “CuKa2.lam” emission profile was used with updated values of CuKα 1 (1.5405929 Å) and Kα 2 (1.5444274 Å) from Table IV of Hölzer et al. (Reference Hölzer, Fritsch, Deutsch, Härtwig and Förster1997). Phase compositions of the jet-milled films and samples synthesized by the thermal reaction of these powders (Table II) were estimated using the known crystal structure of each phase in converged whole-pattern refinements with TOPAS.
The phase compositions are estimated values because the anisotropic thermal parameters for Ag3O have not been published and could not be reliably refined; they were approximated using isotropic parameter B eq = 1 for oxygen and silver. Furthermore, the phase compositions are estimates because the nature of point defects and their effect on site occupations is unknown for Ag3O and Ag2O. Figures of the refined patterns from TOPAS include color-coded tick marks to indicate the hkl angles for each phase corresponding to the color-coded empirical formula shown in the legend.
For powder patterns collected as a function of temperature and time, the initial values of refined variables were taken from refinements of patterns adjacent in time, forward and backward, except for the first pattern collected in the in situ experiment which has initial values based on all of the refinements in the study. The uncertainty of phase abundance was qualitatively estimated from the typical variations of wt% observed when slightly different initial values and sequences of refining the same set of variables resulted in convergence with similar goodness of fits (GoFs).
For each experiment, the 0° 2θ angle was either measured or refined. Sample displacement was refined as a function of cosθ for patterns collected in Bragg–Brentano configuration and as a function of sinθ for patterns collected in transmission geometry. The PV_TCHZ peak-profile function was used with the variables u, v, w, and x either refined or determined from SRM 640e Si and SRM 660a LaB6 refinements. The profile variables y and z were fixed at zero. The crystal size and strain were refined using Lorentzian and Gaussian profiles as needed for the best fit. Diffuse scattering was modeled using a “peaks phase” with a profile selected to provide the best fit to the Bragg peaks. The background signal was modeled using a 3rd-order Chebychev and 1/X functions.
D. Ball milling parameters and ex situ heating
An MM 200 Retch Mixer Mill was used to ball mill film 1 (B1) at 30 Hz for 90 min in air at 50% RH with an approximately 100 mg sample spiked with a couple wt% SRM 640d Si. The mill and sample were noticeably warmed by milling; their temperatures were not monitored. After milling, the mill was allowed to cool for 2 h before the specimen was transferred to ambient temperature. The entire sample was heated ex situ to 80, 100, 120, and 150 °C in air and cooled to ambient at ≤ 1 °C min−1 after each heating. XRPD patterns were collected after each heat treatment at ambient conditions.
E. Sonication parameters
Approximately 0.5 g of jet-milled film 1 was sonicated at room temperature in a 4 ml suspension with isopropyl alcohol (IPA) for 45 s, and 1 g of jet-milled film 1 was sonicated in a 10 ml suspension with cyclohexane for 3 min. The sonication experiments used a Cole Parmer model CV18 probe operated between 6 and 12 W at ambient conditions.
III. RESULTS
The thermal-reaction products of jet-milled film 1 were analyzed in situ and ex situ. The thermal-reaction products of films 2 and 3 were analyzed ex situ in an ambient atmosphere. The amount of Ag2CO3 in the jet-milled films was determined by potentiometric acid-base titration to be approximately ≤10 wt% depending on the time (t) and temperature (T) of exposure to ambient air. The identification and phase compositions of the three reactant films are provided in Table I; these compositions excluded Ag2CO3 and were normalized to 100%. The identification, reaction conditions, and phase compositions of the synthesized samples are provided in Table II.
A. In situ thermal reaction of film 1 from 25 to 148 °C
In situ XRPD patterns were collected on jet-milled film 1 heated from 25 to 148 °C in N2 flowing at 100 standard cm3 min−1 (Figure 2). The temperature-time course for the first part of the experiment is shown in Figure 3; heating continued at 130 °C for 25.5 h and 148 °C for 18 h and then 39 h. A brief decrease in temperature to 102 °C occurred between the 6.5 and 25.5 h holds at the 130 °C target over a 2-min period. Patterns were collected over a 2θ range from 30 to 59° for 15 min at the end of each temperature hold. The ramp rate between temperature holds was 2 °C min−1.

Figure 2. (Color online) Overlays of in situ XRPD patterns of film 1 heated in N2 from 25 to 150 °C.

Figure 3. The temperature-time course for the thermal reaction of film 1 in N2 from 25 to 130 °C. After the 6.5-hour hold at 130 °C heating continued at 130 °C for 25.5 h, and then 150 °C for 18 and 39 h.
The estimated abundance of phases as a function of temperature is shown in Figure 4 starting at 80 °C, just before the reaction of Ag2O and Ag starts to be detected at 90 °C. The lines through the data points are provided for visual guidance. The abundances for samples heated ≤80 °C were the same as the abundances at 80 °C, within the estimated uncertainty of ± 2 wt%.

Figure 4. The estimated phase abundances as a function of temperature for the in situ thermal reaction of film 1 heated in N2.
Pawley refinements for patterns at 25, 80, and 120 °C are shown in Figure 5. The GoFs for all refinements of the in situ reaction products ranged from 1.1 to 1.35; R wp ranged from 3.7 to 5.1. Diffuse scattering under the strong Bragg peaks was present in all of the patterns and was modeled as a “peaks phase” with maxima near 34.0, 37.5, and 44.0° displayed below the XRPD pattern. The unit-cell length of Ag2O as a function of temperature, a(T), is displayed in Figure 6 along with the time for each temperature hold.

Figure 5. (Color online) Pawley refinements for film 1 heated in N2 at 25, 80, and 120 °C.

Figure 6. Nonstoichiometric Ag2O a(T) for the in situ reaction of film 1 heated in N2. XRPD patterns were collected for the last 15 min of each hold period.
B. In situ thermal-reaction kinetics of film 1 at 100 and 130 °C
The thermal reaction of jet-milled film 1 was analyzed in situ near a relative humidity of 100% at 100 °C in N2 as a function of time (Figure 7). The estimated phase abundances expressed as mol% are shown in Figure 8. In situ patterns of another sample of film 1 were also collected every 41 s for 42 h at 130 °C with a ramp rate from ambient of 35 °C min−1 in an ambient atmosphere (Supplementary Figure S5). The unit-cell length of Ag2O as a function of time at 100 and 130 °C is shown in Figure 9.

Figure 7. (Color online) Overlays of in situ XRPD patterns of film 1 heated at 100 °C in N2 for 0, 8, and 15 h.

Figure 8. Phase abundance of film 1 at 100 °C in N2 as a function of time.

Figure 9. Nonstoichiometric Ag2O a(t) for the in situ reaction of film 1 at 100 and 130 °C in N2.
C. Ag3O unit-cell parameters as a function of temperature
The Ag3O unit-cell volume as a function of temperature, V(T), for the in situ reaction of film 1 is shown in Figure 10 along with the Ag3O-rich (78 wt%) product of the ex situ thermal reaction (rxn) of film 1 at 60 °C in air for 1 year (product 1e). The quadratic regression is for product 1e, and the numbers indicate the sequence of the analyses.

Figure 10. Ag3O unit-cell V(T) for the in situ thermal reaction of film 1 in N2 starting at ambient temperature and for the in situ cooling and heating of product 1e. The time sequence of the cooling and heating of product 1e is indicated by the number next to the data point.
Expressing the V(T) for Ag3O and Ag as a percentage difference of the unit-cell volume at 25 °C allows a relative comparison of their thermal expansion for the in situ experiments on film 1 and product 1e (Figure 11). The regression for Ag3O in Figure 11 was done using data points 1, 2, and 3, and a quadratic function in case data point 4 near 200 °C was decreasing as a function of time during the ramp from low temperature. A decrease in volume with time is clearly observed for point 5. The Ag3O a and c axes for these two experiments as a function of temperature are shown in Figure 12. The dashed and continuous lines are regressions on a and c, respectively, for product 1e.

Figure 11. V(T) for Ag3O and Ag as a percentage difference of their unit-cell volumes relative to 25 °C for the in situ thermal reaction of film 1 and the ex situ, thermal-reaction product 1e. The forward-time sequence for product 1e is indicated by the number next to the data point.

Figure 12. The a axis and c axis of Ag3O as a function of temperature for the in situ thermal reaction of film 1 heated in N2 and the in situ, cooling and heating of thermal-reaction product 1e. The regression lines are for the cooling of product 1e and are extrapolated to higher temperatures.
The jet-milled films were also reacted ex situ at temperatures between 40 and 141 °C in air for up to 1 year (Table II). The cooling rate for these experiments was only controlled for samples 3a, 3b, and 3c, which were cooled with the same procedure; their cooling rate was unavailable. The Ag3O c/a(T) is shown in Figure 13 along with the c/a for the in situ thermal-reaction experiment described in section III.A.

Figure 13. The c/a (T) for Ag3O produced by in situ and ex situ thermal reactions. The numbers next to the markers for product 1e indicate the time sequence of the analyses.
The c/a for the in situ reaction of film 1 from 40 °C to the first 100 °C hold have significantly more uncertainty compared to the c/a from the second 100 °C target to the 150 °C target where the signal from Ag3O was much higher and the uncertainty of a and c was much less. The relatively high scatter of c/a values is from propagation of errors in c and a combined with the poor reproducibility of determining the position of very weak, convoluted, broad peaks. These data points are graphed with gray circles in Figure 13. The GoFs of these refinements were 1.15 ± 0.05, and the Durbin Watson d statistic for serial correlation was 1.7 to 1.8, which indicates no strong correlations. The abundances of phases determined in these Pawley refinements varied by about ± 2 wt% for each phase.
D. Ex situ thermal reactions
The ex situ thermal-reaction product ID, reaction temperatures, reaction times, reaction atmospheres, and estimated phase compositions are provided in Table II. Ag3O c/a vs. V for the ex situ reaction products at ambient temperature, 24–29 °C, is shown in Figure 14 along with the values from Beesk et al. (Reference Beesk, Jones, Rumpel, Schwarzmann and Sheldrick1981) scaled by the weighted-average MoK  $\bar{\alpha }$ wavelength (0.710782 Å) calculated using the wavelengths of MoKα 1 and Kα 2 and their relative intensities from TOPAS (MoKa2.lam) to update the MoKα = 0.71069 Å used by Beesk et al. (Reference Beesk, Jones, Rumpel, Schwarzmann and Sheldrick1981); the scale factor = 0.710782 ÷ 0.71069 = 1.000130 and was multiplied by their published unit-cell lengths. The published and scaled values of c/a from Beesk et al. (Reference Beesk, Jones, Rumpel, Schwarzmann and Sheldrick1981) are shown as gray and black squares separated by an arrow, respectively. The c/a for 3c was, within the uncertainty of the refinement, the same as for 3a; it was not graphed in Figures 13 and 14 for clarity. Several other reaction products listed in Table II were not included in the graph because the angle range and resolution of their patterns was too limited for a reliable unit-cell refinement relative to those graphed.
$\bar{\alpha }$ wavelength (0.710782 Å) calculated using the wavelengths of MoKα 1 and Kα 2 and their relative intensities from TOPAS (MoKa2.lam) to update the MoKα = 0.71069 Å used by Beesk et al. (Reference Beesk, Jones, Rumpel, Schwarzmann and Sheldrick1981); the scale factor = 0.710782 ÷ 0.71069 = 1.000130 and was multiplied by their published unit-cell lengths. The published and scaled values of c/a from Beesk et al. (Reference Beesk, Jones, Rumpel, Schwarzmann and Sheldrick1981) are shown as gray and black squares separated by an arrow, respectively. The c/a for 3c was, within the uncertainty of the refinement, the same as for 3a; it was not graphed in Figures 13 and 14 for clarity. Several other reaction products listed in Table II were not included in the graph because the angle range and resolution of their patterns was too limited for a reliable unit-cell refinement relative to those graphed.

Figure 14. The Ag3O c/a at room temperature for the products of ex situ thermal reactions, ball milling, and sonication reactions as a function of unit-cell volume. The data from Beesk et al. (Reference Beesk, Jones, Rumpel, Schwarzmann and Sheldrick1981) (gray square) were measured on a single crystal hydrothermally synthesized on Ag metal and were scaled using MoK  $\bar{a}$ from TOPAS as shown by the arrow from the gray square to the black square.
$\bar{a}$ from TOPAS as shown by the arrow from the gray square to the black square.
The a axis versus c axis at ambient temperature is shown in Supplementary Figure S7. Some of the whole-pattern refinements are shown in Figures 15 and 16. The very weak CuKβ transmitted by the multilayer mirror for the sharp, strong Ag3O 111 is located at 34.5°; this peak was not included in the refinements because it did not significantly affect the residual.

Figure 15. (Color online) TOPAS Pawley refinements for products 1d and 3a. The color-coded tick marks correspond to the color-coded formulas for Ag3O and Ag.

Figure 16. (Color online) TOPAS Pawley refinements for ball-milled film 1 and product 2a. The color-coded tick marks correspond to the color-coded phases detected.
E. Ball-milled jet-milled film
The ball-mill experiment produced an estimated 76 wt% of Ag3O in sample B1 with broad peaks accounted for by an average size (coherence length) and microstrain (ε 0) (Figure 16). SRM 640d Si peaks in the pattern were used to define the peak-profile parameters for instrumental resolution. Relatively weak, very broad peaks for the LT Ag2CO3 polymorph (Norby et al., Reference Norby, Dinnebier and Fitch2002) were Pawley refined with an average coherence length = 267(30) Å and ε 0 = 0.0026(1) using a Lorentzian profile. Ag2O was not detected.
The average coherence length of the Ag3O in B1 was 280(2) Å; only a Lorentzian profile component was significant. ε 0 was 0.00097(1); only a Gaussian profile was significant. The steep rise of intensity at low angles was small-angle scattering (SAXS) (Figure 16). The origin of the SAXS signal was not investigated and not recorded for most of the patterns in the present study.
Ex situ heating of B1 to 80, 100, and 120 °C in air each linearly decreased the Ag3O unit-cell volume and abundance (Figure 14; Supplementary Figure S7). On heating to 80 °C, the a axis decreased while the c axis was not significantly different (Supplementary Figure S6), and the SAXS signal decreased by about 80%. Heating to 100 and 120 °C decreased the c axis with essentially no change in the a axis (Supplementary Figure S7). On heating these samples, the coherence length of B1a, B1b, and B1c was 351(5), 395(4), and 479(8) Å, respectively, and ε 0 was 0.0007(1), 0.0006(1), and 0.0005(1), respectively. After 5 days at 150 °C, only Ag metal was detected.
F. Sonicated jet-milled films
The reaction of sonicated films produced an XRPD pattern of Ag3O with sharper peaks than the Ag3O peaks from the ball-milled product. LT Ag2CO3 has broader Bragg peaks than the Ag3O. An estimated <1 wt% of Ag2O remained after both sonication reactions. The products of the cyclohexane sonication for 3 min contained more Ag than the products from sonication in IPA for 45 s (Table II). This might reflect the more reducing nature of cyclohexane compared to IPA.
The Ag3O c/a for the material synthesized in ball milling and sonication experiments are graphed in Figure 14 but not included in the linear regression because of the strong overlap of several strong peaks for Ag3O and Ag2CO3 (Supplementary Figures S8 and S9) which could affect the accuracy of their unit-cell parameters. The peak broadening for Ag2CO3 was significantly anisotropic; this was not included in the structural model. Therefore, the unit-cell parameters of Ag2CO3 could be less accurate compared to the parameters from the Pawley refinements of the patterns displaying very sharp peaks of Ag3O with mostly instrumental and isotropic broadening.
IV. DISCUSSION
A. Diffuse scattering: Ag2CO3 and stacking faults in Ag
Diffuse peaks were used as background in the whole-pattern refinements to facilitate the best fit to the Bragg peaks and maximize the accuracy of the unit-cell parameters. One of the diffuse peaks ranged between 42 and 45° (Figure 5) and was used to fit the asymmetry of the Ag 200 hkl which has a low-angle tail. The tail is a consequence of a reciprocal-lattice intensity rod generated by stacking faults (Schields et al., Reference Schields, Dunwoody, Mamak, Gendron and Bates2007). A characteristic peak generated by stacking faults in Ag starts at about 36° and extends several degrees to lower angles; this saw-toothed peak was too weak to detect. The Ag stacking-fault probabilities for many patterns in the present study were refined using the Warren model in MAUD 1.999 (Lutterotti et al., Reference Lutterotti, Matthies and Wenk1999) which accounts for the 200 tail. The probabilities were in the range of several tenths of a percent.
Weak, diffuse scattering with a maximum ranging from 33.7 to 34.1° was present in all of the Ag3O-rich patterns. The width (FWHM) of this maximum decreases continuously as a function of temperature (Supplementary Figure S10) for the in situ thermal reaction of film 1 and products 1d and 3b. The net intensity of this maximum is approximately constant as a function of temperature for the in situ thermal reaction of film 1. If this peak was from thermal-diffuse scattering by acoustic phonons in Ag3O, we expect it would increase its intensity with increasing temperature. We hypothesize this peak is from metastable β Ag2CO3, one of the high-temperature forms, which has two relatively intense peaks very near the position of this diffuse-scattering maximum that merge into one peak with a maximum of 33.76° when broadened. The next strongest peaks for β Ag2CO3 are near 19.34 and 48.51°. The Pawley fit of the pattern of the ball-milled sample, which was rich in LT Ag2CO3, required weak peaks near these three presumed β Ag2CO3 peak positions. However, the peaks from β Ag2CO3 are much too weak and broad to determine the unit-cell parameters. Norby et al. (Reference Norby, Dinnebier and Fitch2002) determined its unit-cell parameters at 180 °C.
Another very diffuse peak centered at ~37.5° is included in most of the Pawley refinements on patterns of samples containing at least a minor amount of Ag2O. Ag2O is known to readily form Ag2CO3 when exposed to an atmosphere containing CO2; however, crystalline Ag2CO3 was only detected in trace amounts in the thermal-reaction products. We hypothesize Ag2CO3 forms a very thin layer on Ag2O (Culbertson, Reference Culbertson1964) starting at ambient conditions, and, unless the layer is removed, the rate of formation decreases with time. Fresh Ag2O surfaces were being created during the ball-mill and sonication experiments, which produced abundant crystalline LT Ag2CO3 presumably from reaction of Ag2O and ambient CO2. We are unsure of the identity of this peak.
B. Thermal reaction of jet-milled films
All of the phases in film 1 displayed broad Bragg peaks indicating the presence of disorder, which was modeled using crystallite size and microstrain. The values of size and strain are not meaningful because of the short range of 2θ and the inaccuracies of refining very broad, weak peaks at higher angles. The parameters of size and strain facilitated the best fit to the Bragg peaks.
The abundances of the phases remained constant until ~80 °C (Figure 4); at 99 °C, the abundance of Ag3O significantly increased from ~8 to ~50 wt%. From 129 to 148 °C, the amount of Ag3O begins to decrease, and the amount of Ag increases; this indicates Ag3O is decomposing to Ag. The Ag3O unit-cell volume decreases with time at 148 °C (section IV.D.1) while decomposing to Ag.
The kinetics and extent of the thermal reaction of Ag and Ag2O depend on the heating rate. For example, if the temperature is increased at 35 °C min−1 from ambient temperature to 130 °C, only the initial 8 wt% Ag3O with sharper peaks is detected after 41 h. Other examples of the dampened reaction with relatively high heating rates were also observed.
C. Kinetics of the thermal reaction to form Ag3O at 100 and 130 °C
The estimated phase abundance of film 1 heated to 100 °C in N2 as a function of time (Figure 7) shows the reaction rates are constant (Figure 8). The Ag2O reaction rate, –1.003 mol% h−1, is 68% faster than Ag, −0.597 mol% h−1. For stoichiometric Ag2O and Ag3O, the reaction rate for Ag and Ag2O to form Ag3O must be equal because the reactants combine in equimolar ratio. This difference in reaction rates is readily apparent in the Ag 200 peak, which decreases intensity very little compared to the Ag2O 111 peak as the reaction proceeds (Figure 7); this can also be seen in Figure 2.
The difference in reaction rates indicates nonstoichiometry in Ag3O and/or Ag2O. For example, if Ag2O is oxygen deficient and Ag3O is stoichiometric, its formula would be Ag2O1−x with x = 0.17 to balance the reaction. If Ag2O is stoichiometric, then the silver suboxide would be oxygen rich, Ag3O1+x with x = 0.15. Linear combinations of these compositional end members are possible. Analogous examples could be cast with a variable silver stoichiometric coefficient and stoichiometric oxygen content.
Both film 1 and film 3 reactants started with about the same Ag/Ag2O molar ratio of two. If the rates are constant for the thermal reactions of these films, Ag2O would be depleted in 34 h with 17.5 wt% remaining Ag and 82.5 wt% Ag3O produced. These predicted amounts are very close to the amounts measured with ex situ thermal-reaction experiments at 100 °C shown in Table II for products, 1b (48 h), 3a, and 3b: 84, 83, and 81 wt% Ag3O, respectively. This observation indicates the constant reaction rate largely persisted in 1b until the limiting reactant Ag2O was almost depleted. An almost constant rate indicates the reactants remain readily available to each other, the reactivity of the reactants remains constant, and diffusion is not limiting the reaction.
The kinetics and extent of the thermal reaction of Ag and Ag2O depend on the heating rate. The thermal reaction was not detected in film 1 after ramping to 130 °C at 35 °C min−1 (Supplementary Figure S5). The only change after 41 h was the sharpening of the Ag2O and Ag3O peaks.
D. Ag3O unit-cell parameters
1. V(T)
The unit-cell volume of the starting disordered Ag3O in film 1 increased with about the same slope (Figure 10, dotted line) as the ambient to low-temperature linear slope of V(T) for product 1e extrapolated to higher temperatures (Figure 10, dashed line). From 119 to 148°, the Ag3O unit-cell volume of film 1 increases with about the same slope as the linear regression for the low-temperature expansion of product 1e extrapolated from 25 to 122 °C (Figure 10, dashed line). This volume expansion of product 1e is significantly different than the quadratic regression of the low-temperature expansion of film 1 (Figure 10, solid line). However, sparse data points over these ranges preclude the distinction between linear and quadratic volume expansion of Ag3O in both samples.
Figure 11 compares the volume expansion of Ag3O and Ag measured in these same in situ experiments of film 1 and product 1e. The expansion of Ag3O is about 80% less than Ag over the entire measured range. The close agreement of the Ag volumes measured in these separate in situ experiments indicates the determined unit-cell parameters have repeatability adequate for comparison between experiments. Suh et al. (Reference Suh, Ohta and Waseda1988) showed their measured thermal expansion of Ag, Au, Cu, and Ni by XRPD and dilatometry from 25 °C to at least 1000 °C was modeled well by a quadratic equation with positive curvature, and their results were consistent with the data from several previous studies. Many closest-packed structures have thermal expansion with positive curvature (Iwanaga et al., Reference Iwanaga, Kunishige and Takeuchi2000). Distinguishing between linear and quadratic functions for the thermal expansion of Ag3O requires many more data points throughout the temperature ranges of interest than were collected in the present study.
The volume of Ag3O in film 1 decreases 0.2% from 79 to 94 °C when the thermal reaction of Ag and Ag2O starts to form Ag3O. This volume decrease could be due to the Lorentz effect described by Öztürk et al. (Reference Öztürk, Yan, Hill and Noyan2015) if the size of the crystal prior to the thermal reaction was < 100 Å and the size increases from 79 to 94 °C. The short 2θ range, broad peaks, and weak signal preclude the refinement of size and microstrain for patterns in this temperature interval.
2. a(T) and c(T)
Figure 12 shows the Ag3O a axis and c axis expansions from ambient to low temperatures display a slight positive curvature as reflected in its volume expansion. Their expansions are anisotropic with the a axis slope and absolute change about twice those of the c axis from −162 to 25 °C. The lengths of the a and c axes show an abrupt decrease from 80 to 95 °C as expected from the corresponding abrupt volume decrease. The c axis then continues to increase with about the same or slightly increased initial slope while the a axis is approximately constant as temperature rises, a striking difference.
The c/a(T) for Ag3O produced by in situ thermal reactions (Figure 13) shows a negative, linear slope (−8.163 × 10−6 C−1) for T ≤ ambient. This slope is compared to the c/a(T) slope of −2 × 10−5 C−1 for Ag2F (Williams, Reference Williams1989), which has a structure similar to Ag3O except the octahedral interstices in the cp Ag bilayers are all occupied by F in every other adjacent bilayer. The linearity of c/a(T) is a consequence of a frozen-in anisotropy of a (T) and c(T). The inversion of c/a(T) near ambient T reflects the decrease in the expansion of the a axis, while the expansion of the c axis continues to increase with increasing temperature above ambient T (Figure 12). Between 194 and 293 °C, the Ag3O in product 1e completely transformed to Ag and Ag2O.
3. c/a vs. V at ambient T
The c/a ratios vs. volume (c/a vs. V) at ambient T for the ex situ thermal-reaction profucts (Table II), ball-milling product, and sonication products are shown in Figure 14. The linearity of c/a vs.V for the thermal-reaction and sonication products indicates a dependence on a compositional variable and/or variable state of disorder; both are expected to change c/a vs. V. We presume the single-crystal studied by Beesk et al. (Reference Beesk, Jones, Rumpel, Schwarzmann and Sheldrick1981) was in equilibrium with Ag during their several-day, hydrothermal synthesis. The phase composition of the products reveals samples with <3 wt% Ag and excess Ag2O (Ag poor), and a Ag-rich group with excess Ag and almost no detectable Ag2O as encircled in Figure 14. This trend suggests the redox potential associated with solid-solid contacts is a factor determining the position along the linear relation.
Ag-rich phase compositions of the reaction products are expected to be most reducing and associated with oxygen-poor nonstoichiometric Ag3O with a relatively smaller unit-cell volume. Ag-poor compositions are expected to be relatively oxidizing and associated with oxygen-rich nonstoichiometric Ag3O with a relatively larger unit-cell volume.
The heating and cooling rates of samples 3a and 3b were controlled to be the same; the atmosphere was air for both. The only difference between the thermal-reaction conditions for this pair is the one-week longer time at 100 °C for 3b. We hypothesize the smaller volume and increase of c/a of 3b relative to 3a was caused by the loss of oxygen rather than a possible change in order and disorder generated by temperature. If this hypothesis is correct, the increase of c/a (V) could be extrapolated to explain the inversion of the c/a (T) above ambient (Figure 13) as a loss of oxygen with increasing temperature in the in situ thermal reaction of film 1. The same reasoning applies for points 4 and 5 observed for the heating of product 1e where the c/a increases and unit-cell volume decreases with time at 194 °C.
a Ex situ heating of ball-milled sample
The c/a (V) for the ball-milled samples (Figure 14) are significantly removed from those fit with a linear function, and they display broad Bragg peaks. The broadening was mostly generated by short coherence lengths (crystal size) rather than microstrain. The other samples in Figure 14 all have sharp Bragg peaks with essentially no intrinsic peak broadening. We consider the possibility of the refined unit-cell parameters being affected by a Lorentz correction (Öztürk et al., Reference Öztürk, Yan, Hill and Noyan2015) which significantly biases the position of Bragg peaks to lower Bragg angles and larger d-space for crystals < 100 Å in size. Our refined estimate of the average coherence length, 225(5) Å, in the ball-milled sample is expected to be too large to cause a significant bias to larger d-spaces and corresponding unit-cell volumes.
The continuous drop in unit-cell volume on heating the ball-milled sample and the corresponding linear increase of the Ag content is consistent with Ag3O decomposing to Ag metal by volatilizing oxygen. The volume change during the first heating at 80 °C was almost completely from the decrease in the a axis. Subsequent volume changes at 100 and 120 °C were mostly from a decrease in the c axis. The increase of size and decrease in microstrain with increasing temperature-time treatments is consistent with crystal growth and a decrease in disorder. However, these microstructural changes are relatively small.
E. Ag2O a(T) and a(t)
The Ag2O a(T) (Figure 6) shows the effect of negative thermal expansion (Tiano et al., Reference Tiano, Dapiaggi and Artioli2003) and kinetic changes in composition due to nonstoichiometry and thermal equilibration. The almost flat to negative change at the start of heating from ambient and the 1.7 h hold at 120 °C reveal the negative thermal expansion because the kinetics of compositional changes for these temperatures and hold times are slow relative to the other temperature/time steps.
The large 0.57% expansion of volume over the range shown in Figure 6 is hypothesized to be driven by a decrease in nonequilibrium volume-contracting point defects, which impart nonstoichiometry. Equilibration of these point defects is kinetic as shown in Figure 9 at 100 and 130 °C. The expansion rate from 65 to 100 °C is fast enough to mask the decrease of a from thermal expansion. After the first 100 °C hold, the volume increases much slower. We have characterized this equilibration in Ag2O made with significantly different processes including powders made from as-removed thin films in the present study without jet milling and an as-deposited Ag–Ag2O magnetron-sputtered thin films (Figure 17).

Figure 17. (Color online) Ag2O a(T) from several studies and the present study. The values for Taylor et al. (Reference Taylor, Omotoso, Wiskel, Mitlin and Burrell2005) were graphed at their annealing temperatures by adding the thermal expansion determined by Tiano et al. (Reference Tiano, Dapiaggi and Artioli2003).
The expansion of Ag2O with increasing temperature was characterized ex situ by Faivre (Reference Faivre1940), Allen (Reference Allen1960), Kato and Anju (Reference Kato and Anju1972), and Taylor et al. (Reference Taylor, Omotoso, Wiskel, Mitlin and Burrell2005) (Supplementary Figure S12). Ag2O films synthesized with oxidizing conditions by Wei et al. (Reference Wei, Mao, Ortiz and Sadoway2011) were determined to have oxidation states of Ag higher than 1+ by X-ray photoelectron spectroscopy, and the corresponding point defects were especially volume contracting (Figure 17).
The Ag–Ag2O thin film studied by Taylor et al. (Reference Taylor, Omotoso, Wiskel, Mitlin and Burrell2005) was synthesized with conditions similar to the thin films of the present study and was found to have a relatively low volume which increased rapidly with mild heating. The Ag2O in these films decomposes completely to Ag metal by 110 °C. We hypothesize the Ag2O films characterized by Wei et al. (Reference Wei, Mao, Ortiz and Sadoway2011) have excess oxygen. The volume increase from the point defects associated with excess oxygen is more than offset by the significant volume contraction from point defects associated with Ag2+ and Ag3+. These hypothetical point defects are likely to be clusters of excess oxygen and higher oxidation states of Ag.
F. SEM photomicrographs of thermal-reaction products
Supplementary Figure S11(a) shows a scanning electron microscopy (SEM) photomicrograph of Ag3O crystals with some facets produced by the thermal reaction of a jet-milled film. The reaction temperature for this product was between 40 and 100 °C; the time period is unavailable. The smaller irregular particles and warty, faceted crystals imbedded in the larger crystal look like they are merging with the larger faceted crystal. This morphology contrasts the irregular and rough morphologies of the in situ thermal reaction described in section III.A heated to 148 °C for 39 h and product 1e [Supplementary Figures S11(b) and S11(c)].
G. Ag3O tunnel structure
The Ag3O structure determined by Beesk et al. (Reference Beesk, Jones, Rumpel, Schwarzmann and Sheldrick1981) consists of a sublattice of distorted hcp Ag with a two-layer repeat and oxygen occupying two-thirds of the octahedral interstices in every other adjacent cp layers (Figure 1). The Ag in cp layers occupied by oxygen are expanded from their perfect hcp positions, and the Ag6O octahedra have three adjacent, empty octahedral interstices. These empty octahedral interstices share rotated trigonal Ag3 faces along the c axis shown in Figure 1(b) as two equilateral triangles (blue) rotated by 60° around their common center point. The empty octahedra form a continuous, trigonal tunnel of space through the trigonal planar Ag (Ag–Ag 2.86 Å), which are 1.65 Å from the tunnel axis; this “tunnel radius” is slightly larger than the effective radius of 1.40 Å for O2− and the effective radius of 1.445 Å for ccp Ag metal. These empty interstices also form rows coincident with the a axis and c axis in the oxygen-coordinating Ag cp bilayer. The tunnels are a modality for high ionic conductivity of oxygen and silver as well as a readily available diffusion path. The tunnels also provide a connection between the Ag bilayers occupied by oxygen and the Ag bilayers unoccupied by oxygen.
H. Variable oxygen content of nonstoichiometric Ag3O analogs
The crystal structure of Ag3O reported by Beesk et al. (Reference Beesk, Jones, Rumpel, Schwarzmann and Sheldrick1981) was refined using anisotropic thermal-displacement parameters; however, these parameters were not published. These authors do not report they explored the possibility of variable occupancy of oxygen in the ordered octahedral site and the disordered occupation of oxygen in the layer of nominally unoccupied interstices.
Several metal suboxide structures with variable occupation of oxygen in the octahedral interstices of the close-packed metal sublattice are published for Ti, Zr, and Hf (Banerjee and Mukhopadhyay, Reference Banerjee and Mukhopadhyay2007). Hirabayashi et al. (Reference Hirabayashi, Yamaguchi and Arai1973) reported ex situ high-temperature experiments in the Hf–O system with 10–20% atomic oxygen. The unit-cell parameters of these samples were measured with XRPD. For the composition close to the Hf6O stoichiometry and space group P  $\bar{3}$1c, the structure has oxygen in every cp bilayer. They found the unit-cell volume and the c/a increased with increasing oxygen. Using single-crystal neutron diffraction, they determined oxygen has an ordered and variable occupation of octahedral interstices as a function of temperature. When Hf6O was quenched from 1000 °C, the oxygen occupancy was disordered and increased the volume by 0.6% relative to the ordered structure with the same oxygen content.
$\bar{3}$1c, the structure has oxygen in every cp bilayer. They found the unit-cell volume and the c/a increased with increasing oxygen. Using single-crystal neutron diffraction, they determined oxygen has an ordered and variable occupation of octahedral interstices as a function of temperature. When Hf6O was quenched from 1000 °C, the oxygen occupancy was disordered and increased the volume by 0.6% relative to the ordered structure with the same oxygen content.
The Ag3O unit-cell volume of film 1 (Figure 10) decreases by 0.2% from 80 to 95 °C when the reaction to produce more Ag3O begins to be detected. If the Lorentz effect discussed in section IV.D.1 is not a significant cause of this decrease, we hypothesize this volume decrease is due to a change in the amount of oxygen and/or the order/disorder of the oxygen. The disorder could be interstitial oxygen occupying the vacant octahedral interstice of the perfect structure, vacant oxygen in the octahedral interstice of the perfect structure, and disordered oxygen in the octahedral interstices of the Ag bilayers which are all vacant in the perfect structure. The presence of planar stacking disorder of the silver and oxygen cp layers provides many possible scenarios with respect to their influence on the nonstoichiometric composition. These scenarios might also lead to the outlying Ag3O volume produced by ball milling and subsequent heating (Figure 14).
I. Qualitative model for the range of c/a in Ag3O
The suite of Ag3O-rich samples with a linear c/a vs. V is hypothesized to have a continuously variable amount of oxygen in the ordered oxygen layer of the perfect structure. The c/a is an additive measure of distortion generated by oxygen, and V is an additive measure of the total amount of oxygen. We hypothesize the linear relation between c/a and volume in Figure 14 and the inversion of c/a above room temperature in Figure 13 are generated by changes in oxygen composition and disordering.
XRPD is relatively insensitive to the 4.7 wt% oxygen in stoichiometric Ag3O, even from relatively large changes in oxygen composition. This insensitivity combined with a high correlation between thermal-displacement parameters and site occupancy obscures the accurate refinement of these variables using XRPD data. We defer to prospective neutron diffraction analysis, which has high sensitivity to oxygen, to determine the origin of c/a (V) at constant temperature and the inversion of c/a above ambient temperature in Figure 13.
V. CONCLUSION
A suite of nonstoichiometric Ag3O-rich samples was synthesized by mild heating, sonication, and ball milling of jet-milled Ag–Ag2O magnetron-sputtered films in air and N2 at ambient pressure. The phase composition, unit-cell parameters, and reaction rates for thermal transformation of the films indicate Ag3O was produced from the reaction of nonstoichiometric Ag2O and ccp Ag.
The variation of the Ag3O unit-cell volumes and c/a as a function of temperature indicates Ag3O has a range of nonstoichiometric compositions. The nonstoichiometry is likely a consequence of the oxygen content and the ordering of oxygen in the octahedral interstices of the hcp Ag sublattice. Ag3O is metastable and decreases its unit-cell volume during kinetic decomposition to Ag when heated above ambient temperature in air and nitrogen.
The thermal volume expansions of Ag and Ag3O below room temperature have similar positive curvature. The relative thermal expansion of Ag3O is less than Ag by about 80% at room temperature and below. The thermal expansion of Ag3O is anisotropic; expansion along the a axis is about twice the expansion along the c axis. This results in a decrease of c/a with increasing temperature below room temperature. Above room temperature, the c/a(T) makes a remarkable inversion of slope due to the decrease of the a-axis expansion to near zero and an increase in the c-axis expansion. The suite of nonstoichiometric Ag3O samples displays a linear relation between c/a and unit-cell volumes at room temperature. The linear relation is hypothesized to be partly controlled by the redox potential of the system and oxygen content of Ag3O. The site occupancy of oxygen in Ag3O is most suited to neutron diffraction analysis.
SUPPLEMENTARY MATERIAL
The supplementary material for this article can be found at https://doi.org/10.1017/S0885715620000561.
ACKNOWLEDGEMENTS
The authors acknowledge Stephan X.M. Boerrigter and Patrick A. Tishmack, AMRI WLF, for review and discussion.





















