INTRODUCTION
Rare earth metals such as Er hold interest for hydrogen storage applications. The ground-breaking work on the erbium-hydrogen system by Lundin (Reference Lundin1968) documents three possible phases (α,β,γ) for this metal hydride. The α phase can be understood as having the same hexagonal symmetry as that of Er metal, but with the addition of hydrogen into the host Er lattice as a solid solution. The β phase has a fluorite-type structure and usually occurs near the formula ErH2; hence, it is also referred to as the dihydride phase. Lundin (Reference Lundin1968) also documented a tri-hydride phase (γ) which has also been recently explored by Tewell and King (Reference Tewell and King2006). As our interest in this study was monitoring the formation of ErD2 from Er metal, we shall limit discussion in this report to the α and β phases.
Synthesis of the β phase via systems such as a pressure-composition-temperature (PCT) apparatus (Ferriz et al., Reference Ferrizz, Louck and King2007) results in powders and films that traverse the phase diagram for the erbium-hydride system. However, many of the synthesis protocols are based on empirical observations of the final synthesized compounds and little is understood regarding the crystallographic behavior of the α and β phase during hydrogen (i.e., protium, deuterium, and/or tritium) loading. As a means to probe the α and β phases while they are forming, we have undertaken in situ deuterium loading with simultaneous neutron scattering. We loaded Er using D2 gas as the scattering length for deuterium (6.671 fm) is of the same order as that of the Er species (7.79 fm), which means that a neutron scattering measurement will give nearly equal sensitivity to the two atomic species, opening up opportunities to monitor structural parameters including site occupancy of the deuterium atoms within the α and β phases. We present here the details of the experimental setup for such an in situ measurement and document the conversion of Er to ErD2 via this technique.
EXPERIMENTAL
High-purity (99.99%) Er metal was milled into chunks (size range of 100 μm–2 mm) via cryo-grinding. We chose to employ a powder specimen because a random neutron scattering of a loose powder provides a more straightforward analysis of the neutron diffraction data sets. No attempt was made to isolate the Er powder from the air atmosphere prior to loading it into the reaction vessel. Therefore, the Er undoubtedly contained a typical oxide passivation layer on the surface of the individual chunks of the powder sample. Because of the large grain sizes of the powder specimen, the surface-to-volume ratios are dominated by the bulk interior of the Er metal grains; the oxygen in the thin (∼60 Å) passivation layer is very small in comparison to the bulk Er and is considered negligible for this experiment.
The schematic of the experimental setup is shown in Figure 1. Vanadium is effectively transparent to neutrons; this metal was the logical choice for the heating element. Additionally, the heat shields for the heating element were fabricated from thin vanadium foil. A thermocouple (type K) was placed down into the reaction vessel to measure the temperature of the sample during heating. The thermocouple was placed about 1 mm above the powder Er sample. The gas system employed a roughing pump/turbo pump system that could obtain vacuum conditions of <10−7 Torr throughout the entire gas system. The gas system also employed an isolation valve between the in situ reactor chamber and the vacuum. This allowed isolation of the system so that various D2 gas pressures could be applied to the sample in the reactor chamber. Addition of D2 gas into the reactor was controlled by a small needle valve plumbed in-line between the D2 cylinder and the in situ reactor. MKS Baratron pressure gauges were employed to monitor the D2 pressures applied to the in situ reactor.

Figure 1. (Color online) Schematic of the experimental setup for in situ D2 loading of Er metal.
Figure 2 shows the actual setup of the reactor vessel, gas system, and heater prior to installation into the experimental chamber of the high-intensity powder diffraction (HIPD) beam-line at LANSCE. A close-up view of the reactor chamber is shown as well. The actual reactor vessel was fabricated from fused silica (∼7 mm diameter). The reactor vessel was attached to a vacuum flange by a glass-to-metal seal. The flange was subsequently attached to the upper part of the gas system. The entire gas system could be adjusted up-and-down by a gasket at the top plate of the setup. This enabled height adjustment for optimal placement of the specimen in the neutron beam.
Figure 3 shows the entire experimental setup installed into the chamber at HIPD ready for a neutron diffraction analysis. The large neutron scattering chamber (approximately 1 m3) of HIPD is typically evacuated to ∼10−5 Torr in order to reduce scatter and improve counting statistics during data collection. The vacuum conditions of the scattering chamber allowed straightforward use of the vanadium heating element without the concern for oxidation of the vanadium metal during heating for temperature values up to 700°C. Because the fused silica reactor vessel would experience various pressures on the internal walls of the reactor

Figure 2. (Color online) In situ reactor, heater, and gas handling system w/top cover (prior to installation).
while simultaneously experiencing vacuum conditions of the neutron scattering chamber on the exterior reactor walls, a trial run was performed without the use of D2 gas. This was accomplished by simply heating the empty reactor vessel within the large HIPD chamber pumped to 10−5 Torr and maintaining atmospheric pressure (air) in the reactor and subsequent attached plumbing system. Cycling of air and vacuum conditions inside the heated reactor (between 300 and 700°C) showed that the fused silica reactor was robust and easily handled pressure cycling. As our experiment did not plan to exceed 500 Torr D2 pressures, testing to atmospheric pressure (i.e., ∼600 Torr for Los Alamos, NM) was sufficient to establish functionality of the reactor under conditions expected during loading.
Three sets of detector banks (40°, 90°, and 153°) served to obtain diffraction spectra from the sample. Note that because this is a time-of-flight (TOF) experiment, the specimen and detector banks are fixed. This significantly reduces complications of in situ data collection with respect to specimen and detector alignment. The lower angle (40°) banks have higher intensity with moderate resolution over a large range of d-spacing. The higher angle banks (153°) have better resolution

Figure 3. (Color online) Actual experimental setup on the HIPD beam-line at LANSCE.

Figure 4. (Color online) Rietveld refinement of experimental histograms from 40°, 90°, and 153° detector banks. See text for details.
but a more limited range of d-spacing and lower intensity. The 90° banks are intermediate in resolution and d-spacing range, as compared to the 40° and 153° banks, while still yielding good intensity. All detector banks were used in the analysis. Diffraction data were refined using the program GSAS (Larson and Von Dreele, Reference Larson and Von Dreele2000; Toby, Reference Toby2001).
RESULTS AND DISCUSSION
Histograms shown in Figure 4 demonstrate the excellent refinement of experimental data. As one can see, the solid line calculated from the structural model matches well to the observed patterns (plotted as individual+symbols). Note that a region in the 90° and 40° banks was removed due to scatter from the silica reaction vessel. Otherwise, the patterns were free of experimental artifacts. Excellent residual error values (Rp<5%) were typical for our analysis, indicating the appropriateness of the structural models employed in the refinement. The difference patterns, shown at the bottom of each plot, are also employed as a visual gauge for the quality of the fit; in these refinements the difference patterns were essentially flat. This suggests appropriate modeling of the observed histograms. This also lends credibility to the resulting refined parameters.
To obtain an overall picture of the hydration process, a contour map was generated as shown in Figure 5. This plot (employing data from one of the 90° banks) represents the sequential plotting of histograms from the specimen at 450°C. The plot shows how the neutron diffraction pattern changes as various pressures of D2 are bled in from the D2 cylinder. At the onset of the experiment, the Er powder was at vacuum conditions (∼10−7 Torr) and held steady at 450°C. The histogram for Er is plotted at the bottom of the contour plot and is denoted by the label: no D2. The hexagonal Er structure has its major peak (101) at ∼2.7 Å as shown by the bright band along the x-axis in the contour plot. For Figure 5, strong diffraction peaks appear as bright yellow bands on a red background (color online) or as white bands on a black background (black and white version). Over the course of about 20 h, D2 gas was allowed to flow into the reaction vessel; first at 0.1 Torr, then at 1.0 Torr, and finally at 10 Torr and above.
The contour plot shows how the Er pattern slowly expands (peaks shift to larger d-spacings) as deuterium is added into the reactor vessel. Initially, our observations at 0.1 Torr indicated the formation of only the α phase solid-solution.

Figure 5. (Color online) Contour plot illustrating in situ formation of the α and β phases of erbium deuteride via D2 loading at various pressures. Sample held at 450°C during D2 loading. See text for details.
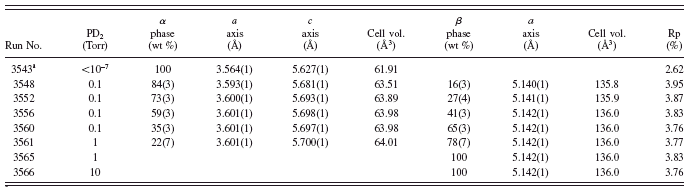
TABLE I. Refined parameters for α and β phases during D2 loading at 450°C.

1 Er metal under vacuum prior to exposure to D2 gas. Parentheses next to numbers indicate 3σ error on the last digit of value.
Saw et al. (Reference Saw, Beaudry and Stassis1983) showed that the α phase for scandium metal has the same structure as the rare-earth metal, but with deuterium locating in the interstitial sites of the hexagonal lattice. We observe this same phenomenon with our Er specimen. The α phase formation is indicated by shifting of the hexagonal peaks, but not the loss of any observed h k l indices. With time, the specimen entered a two-phase region of α phase along with the formation of the β (fluorite) ErD2-x phase. This is clearly distinguished by the growth of the (111) peak of the β phase at ∼3.0 Å d-spacing. This peak is detectable roughly halfway through the 0.1 Torr exposure and grows dramatically with increased D2 pressures. As the reaction continued to progress with time and increased D2 pressure, the β phase became the dominant phase fraction, until the sample fully converted to the ErD2 fluorite as indicated by the strong (220) peak at ∼1.8 Å as well as the (200) and (111) reflections as labeled at the top of the contour plot. We did not detect the presence of Er2O3 in the observed diffraction data. As mentioned earlier, the Er metal most assuredly contained an oxide passivation layer due to the exposure of the metal to air. The lack of detection of Er2O3 would indicate that the passivation layer contributes an insignificant fraction of the measured intensity.
Significant quantification of results could be obtained via Rietveld refinement. Table I documents various refined parameters for the α and β phases during loading. We document our results as a function of run number for purposes of bookkeeping and comparison of the progression of the reaction. However, the run numbers can be roughly translated as time where typical histogram collection runs required roughly 20–40 min. We show the phase fraction (wt%) of the α and β phases as a function of run number in Figure 6. In this plot, the regions are highlighted based on the existing D2 pressure present on the powder during each run. The plot shows that the β phase becomes the majority phase in the reactor even at 0.1 Torr.
The setting of the D2 pressure during analysis was typically performed by adjusting a needle valve controlling the D2 gas until the pressure of the reactor read the desired D2 loading pressure on the MKS Baratron readout. This pressure value tended to decrease over time as the D2 gas was consumed by the Er powder in the reactor. Hence, the experiment required monitoring of the D2 pressure and recurrent adjustments of the needle valve to keep the pressure near to its set-point. We chose to increase pressure to 1.0 Torr between runs 3560 and 3561 because we had reached conditions where the D2 gas pressure of 0.1 Torr stabilized over the Er powder. This may not have been a true thermodynamic equilibrium condition for the sample, as perhaps with more time the sample might have converted fully to ErD2. However, at this point in our experiment it made sense to increase the D2 pressure so that conversion to the β phase could be achieved in the limited time frame of the experimental beam-time.
Upon increase to 1.0 Torr, the conversion to the β phase continued to progress with complete conversion to ErD2 occurring at ∼10 Torr D2 (not shown on plot). Table I shows the refined lattice parameters for both the α and β phases, and one can see the dramatic change of hexagonal lattice with deuterium exposure. In contrast, the β phase shows very little variation in lattice parameter upon formation within the two-phase region as well as when fully converted to ErD2 at 10 Torr. Refinements of site occupancy for the fully converted β phase indicated essentially full tetrahedral (8c) deuterium sites at (1/4 1/4 1/4) for space group F m-3m (225). This suggests that the β phase forms with near stoichiometric fluorite composition even at small phase fractions within the two-phase region. Udovic et al. (Reference Udovic, Rush and Anderson1994) demonstrated possible location of hydrogen in the octahedral (4b) sites of the β-TbH2+x. We did not detect significant deuterium occupancy at the octahedral sites for D2 pressures ≤10 Torr.

Figure 6. (Color online) Weight fraction of α and β phases vs run number for D2 loading at 450°C.
However, subsequent overpressures did indicate partial occupancy of the octahedral sites. Overpressure studies will be discussed in a later publication.
CONCLUSION
In situ neutron diffraction yielded structural information regarding deuterium loading for Er metal at 450°C. The α phase shows dramatic cell expansion upon exposure to D2. The β phase shows little structural change in the (α+β) two-phase region. This suggests formation of the β phase near the ErD2 stoichiometry. No significant octahedral-site occupation was detected for deuterium in the β phase at 450°C for D2 pressures ≤10 Torr.
ACKNOWLEDGMENT
The authors would like to acknowledge the hard work of David Hawn during the integration phase of the reactor and vanadium heater assembly. His contributions greatly simplified the measurement process. The authors also thank Dan Kammler for his preparation of the Er powder via cryo-milling. Sandia is a multiprogram laboratory operated by Sandia Corporation, a Lockheed Martin Co., for the United States Department of Energy’s National Nuclear Security Administration under Contract No. DE-AC04-94AL85000.