I. INTRODUCTION
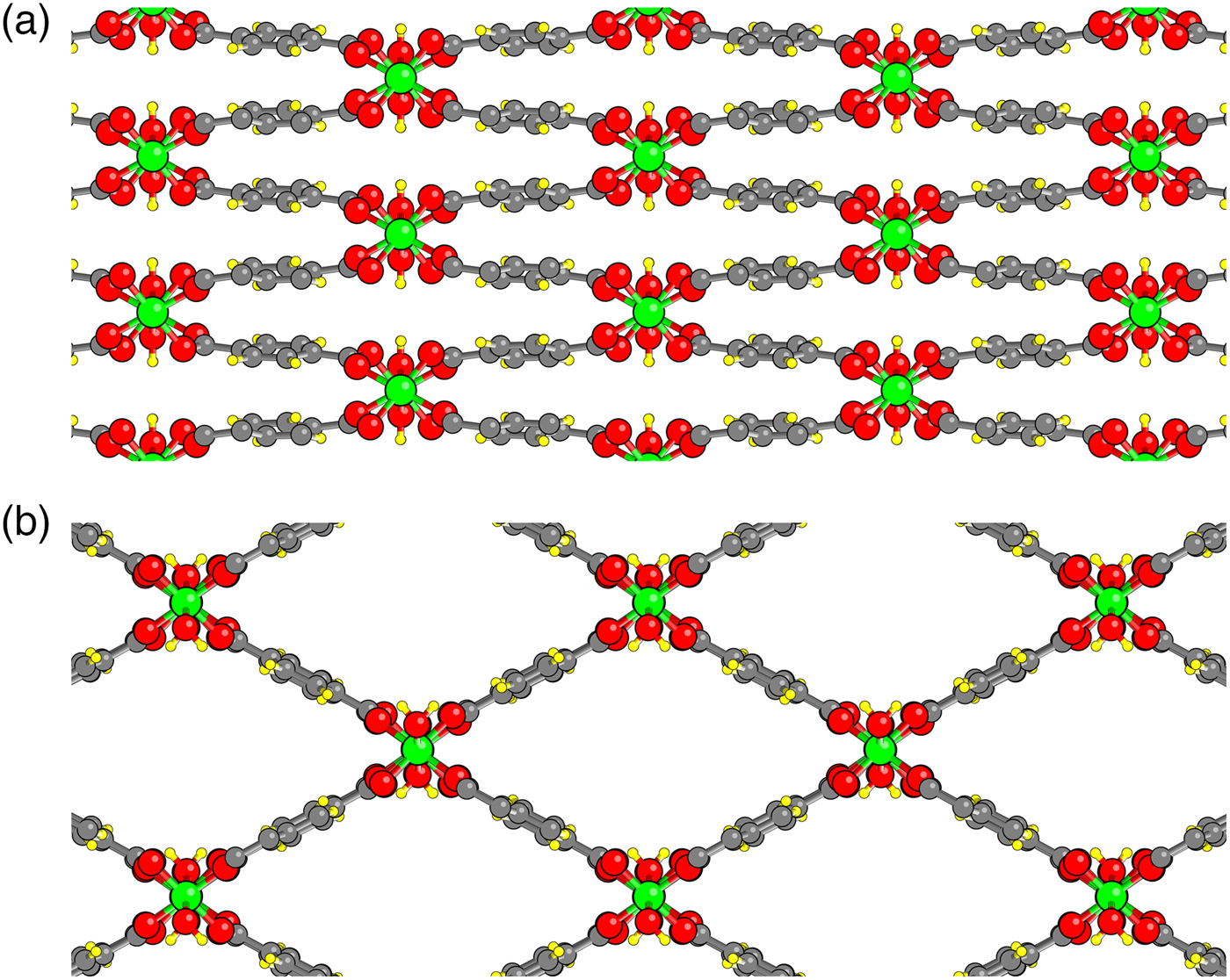
Flexible-framework nanoporous metal-organic materials are fascinating both from a fundamental point of view and for their potential applications such as gas storage, gas separation, sensors, etc. (Férey and Serre, Reference Férey and Serre2009; Alhamami et al., Reference Alhamami, Doan and Chen2014). A well-studied example is the Materials of Institut Lavoisier (MIL)-53(M) family (Serre et al., Reference Serre, Millange, Thouvenot, Noguès, Marsolier, Louër and Férey2002) with formula MC8O5H5, where M is a trivalent species such as Cr, Sc, Al, Ga or Fe (Figure 1). The structure formula can also be written as M(OH)(bdc), where bdc is the (1–4)benzodicarboxylate ion. Each metal ion M is octahedrally coordinated with six oxygens: four from carboxylate groups and two from hydroxyl groups. Zigzag M-OH-M-··· chains are crosslinked by bdc ions to form a space-filling structure having linear pores with a diamond cross-section. These MIL-53(M) compounds typically exhibit both a narrow pore (np) and a large pore (lp) structure, with transitions between the two observed as a function of temperature, pressure, adsorption, etc.
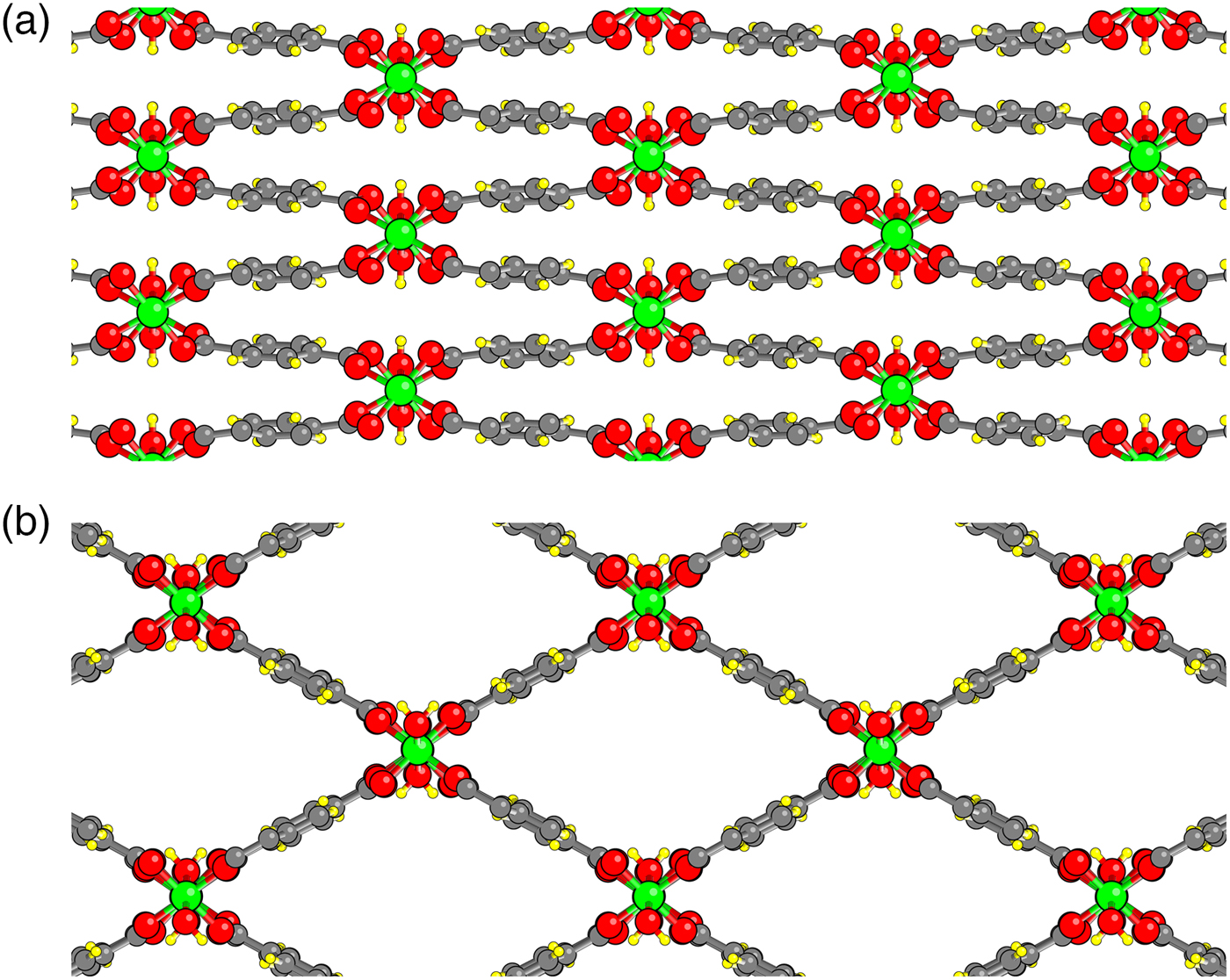
Figure 1. Structure of MIL-53(Cr) viewed down the c-axis. (a) Narrow pore (np) state; (b) Large pore (lp) state. Chromium atoms green; carbon gray; oxygen red and hydrogen yellow.
The adsorption of water into metal-organic frameworks (MOFs) is a topic of great practical interest, as water affects the stability and gas sorption properties of MOFs operating in the atmosphere or in gas mixtures including water vapor (Burtch et al., Reference Burtch, Jasuja and Walton2014; Canivet et al., Reference Canivet, Fateeva, Guo, Coasne and Farrusseng2014; Furukawa et al., Reference Furukawa, Gándara, Zhang, Jiang, Queen, Hudson and Yaghi2014). The MIL-53 family of flexible metal framework materials exhibits an interesting behavior on water absorption, typified by MIL-53(Cr). Dry MIL-53 is in the lp state. As the amount of water adsorption increases above some critical value, MIL-53(Cr) transitions to the np state. At sufficiently high loading, MIL-53(Cr) transitions back to the lp state (Bourrelly et al., Reference Bourrelly, Moulin, Rivera, Maurin, Devautour-Vinot, Serre, Devic, Horcajada, Vimont, Clet, Daturi, Lavalley, Loera-Serna, Denoyel, Llewellyn and Férey2010; Devautour-Vinot et al., Reference Devautour-Vinot, Maurin, Henn and Serre2010). This “breathing” behavior is typical of gas sorption in this family of flexible MOF materials.
The structure of water in MIL-53(Cr) has been studied from a variety of computational and experimental techniques (Llewellyn et al., Reference Llewellyn, Maurin, Devic, Loera-Serna, Rosenbach, Serre, Bourrelly, Horcajada, Filinchuk and Férey2008; Coombes et al., Reference Coombes, Corå, Mellot-Draznieks and Bell2009; Dubbeldam et al., Reference Dubbeldam, Krishna and Snurr2009; Salles et al., Reference Salles, Bourrelly, Jobic, Devic, Guillerm, Llwewllyn, Serra, Férey and Maurin2011; Cirera et al., Reference Cirera, Sung, Howland and Paesani2012; Paesani, Reference Paesani2012; Haigis et al., Reference Haigis, Coudert, Vuilleumier and Boutin2013; Salazar et al., Reference Salazar, Weber, Simon, Bezverkhyy and Bellat2015). For the np phase at a loading of x = 1 H2O per Cr, there is a strong consensus that each water oxygen, or O W, forms a strong hydrogen bond with an H atom of an OH group of the framework. The water hydrogens (HW) may participate in bonding with the framework O atoms or with the O W of other water molecules, but there is less consensus on the details of these bonds. In a powder X-ray structure refinement, Guillou et al. (Reference Guillou, Millange and Walton2011) found a split position for the O W, which they interpreted as evidence for dimerization of consecutive OW via hydrogen bonding.
Haigis et al. (Reference Haigis, Coudert, Vuilleumier and Boutin2013) used ab initio molecular dynamics at the PBE + D2 (Perdew et al., Reference Perdew, Burke and Ernzerhof1996; Grimme, Reference Grimme2006) level to investigate the dynamic behavior of water molecules in MIL-53(Cr) for concentrations x = 1.0 and x = 6.0. For x = 1.0, the np phase was stabilized, each O W remained bound to an OH group via hydrogen bonding, and the H W rotated freely around the dipole axes of the water molecules. For x = 6.0, the lp phase was stabilized, one O W was (quasi)statically bound to each exposed hydroxyl group, while the remaining H2O moved fluidly, although with slower dynamics than in bulk H2O.
While most published density functional theory (DFT) works on molecular adsorption in MOFs to date have been performed at the generalized gradient approximation (GGA) level, it is known (Tran et al., Reference Tran, Stelzl and Blaha2016) that no GGA is excellent for both solids and molecules at the same time, precisely the cases for the MOF and adsorbate molecule, respectively. Tran et al. (Reference Tran, Stelzl and Blaha2016) recommend the use of a meta-GGA (MGGA) density functional for cases involving “both finite and infinite systems”. Additionally, as described in the next section, density functionals at the MGGA level perform better at describing interactions between water molecules than those at the GGA level. With the aim of developing a more accurate description of the geometry and energetics of water adsorption in MOFs, we use a MGGA density functional suitable for water in this work to investigate water adsorption in MIL-53(Cr) for water concentrations of x = 1.0 and x = 1.25.
II. COMPUTATIONAL METHODS
First principles DFT calculations, as encoded in the VASP software (Kresse and Furthmuller, Reference Kresse and Furthmuller1996), were used to calculate the relaxed configurations investigated here and their electronic structures. Certain commercial software is identified in this paper to adequately describe the methodology used. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the software identified is necessarily the best available for the purpose. Cells of composition Cr8O40C64H40(H2O)8x were used for all calculations. The c-axis of the cells used were doubled with respect to that of the conventional cell. Lengthening the repeat distance in the open channels from around 7 Å to around 14 Å allows for a greater variety of water configurations to be studied. Monoclinic np MIL-53(Cr) has the space group C2/c, and orthorhombic lp MIL-53(Cr) has the space group Imma, but in this work we fixed the symmetry at the common subgroup P21 (space group 4) to maintain a monoclinic or higher-symmetry cell while allowing placement of water molecules such that they did not interfere with symmetry-equivalent versions of themselves. The parent C2/c or Imma symmetry is implicit in an ensemble of equal-energy configurations related by translation and other symmetry elements.
It is important to treat the interactions between water molecules properly in DFT calculations (Gillan et al., Reference Gillan, Alfè and Michaelides2016). The widely used “PBE” (Perdew et al., Reference Perdew, Burke and Ernzerhof1996) GGA exchange correlation functional leads to overbonding between water molecules; that is the bonding energy is too high and the hydrogen bonding distances too short; a problem that is even worse when dispersion effects are considered (Santra et al., Reference Santra, Michaelides, Fuchs, Tkatchenko, Filippi and Scheffler2008). Instead, dispersion-corrected MGGA functionals are required to get the proper balance of forces necessary to accurately model water interactions (Pestana et al., Reference Pestana, Mardirossian, Head-Gordon and Head-Gordon2017). In previous work (Cockayne and Nelson, Reference Cockayne and Nelson2015; Cockayne, Reference Cockayne2017), we were able to reproduce benchmarking energies and geometries of small water clusters quite well by using a combination of a PBEsol (Perdew et al., Reference Perdew, Ruzsinszky, Tao, Staroverov, Scuseria and Csonka2008) gradient term, the RTPSS MGGA density functional (Perdew et al., Reference Perdew, Ruzsinszky, Csonka, Constantin and Sun2009; Sun et al., Reference Sun, Marsman, Csonka, Ruzsinszky, Hao, Kim, Kresse and Perdew2011), empirical van der Waals forces at the Grimme D2 level (Grimme, Reference Grimme2006), and a Hubbard U value of 7.05 for the water oxygen, but the combination of parameters that we chose was admittedly ad hoc and not derived self-consistently. In this work, we seek a more transferable density functional, one that has been benchmarked across different systems in the literature. In this regard, we find that good agreement with benchmarking calculations on ice polymorph energies (Brandenburg et al., Reference Brandenburg, Maas and Grimme2015), molecular clusters, and water clusters (Brandenburg et al., Reference Brandenburg, Bates, Sun and Perdew2016) is obtained with the TPSS MGGA functional (Tao et al., Reference Tao, Perdew, Staroverov and Scuseria2003; Perdew et al., Reference Perdew, Ruzsinszky, Tao, Staroverov, Scuseria and Csonka2005) combined with empirical dispersion at the Grimme D3(BJ) level (Grimme et al., Reference Grimme, Ehrlich and Goerigk2011). We note that the parameters of the D3(BJ) dispersion formula were optimized in Brandenburg et al. (Reference Brandenburg, Bates, Sun and Perdew2016) and use their TPSS + D3(BJ) parameterization here. As we did in Cockayne (Reference Cockayne2017), we determined Hubbard correction parameters U and J for Cr within the TPSS + D3(BJ) approach by attempting to fit the unit cell volume and bandgap of corundum Cr2O3. The optimal values found were U = 2.7 eV and J = 1.7 eV.
A 2 × 2 × 2 Monkhorst-Pack k-point grid was used for all calculations. It was verified that calculated energies and forces were converged with respect to the number of k points. The planewave cutoff was 500 eV. In dry MIL-53(Cr), the Cr atoms were found to prefer high spin magnetic states with neighboring Cr in an antiferromagnetic arrangement (Cockayne, Reference Cockayne2017); thus, this magnetic configuration was initialized for the structures studied in this work. Each atomic relaxation was performed at a fixed volume with the cell shape allowed to relax. The volume was varied in small increments to map the enthalpy vs. volume equation of state. In some cases, the water configuration of the MIL-53(Cr) + H2O system spontaneously transformed into a different geometry. In these cases, the new geometry was taken as a starting point for a set of enthalpy vs. volume calculations until crossing points or transformation points between the different configurations were determined.
III. RESULTS
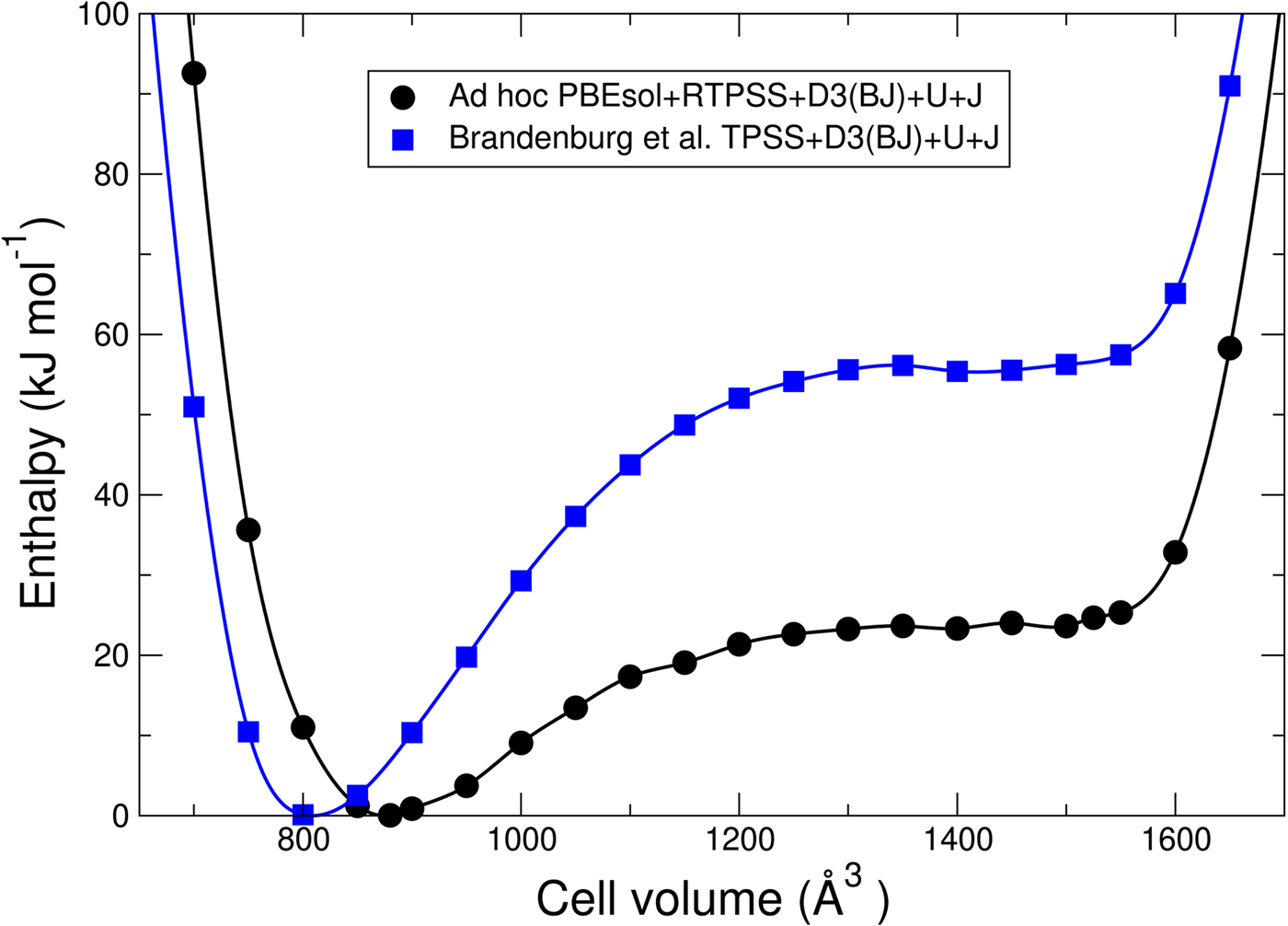
To benchmark our results based on the benchmarked dispersion-corrected MGGA, we first compare the enthalpy vs. volume of dry MIL-53(Cr) using the benchmarked MGGA functional with previous results (Cockayne, Reference Cockayne2017) on MIL-53(Cr) that used the ad hoc MGGA functional described in the Methods section. The results are shown in Figure 2. For comparison with other results in the literature, one mole refers to one mole of Cr4O20C32H20(H2O)4x throughout this work, and cell volumes are given for the “primitive” cell of this composition. Both sets of calculations have a minimum enthalpy for the np phase in contrast to experimental results showing that the lp phase is stable at all temperatures. In Cockayne (Reference Cockayne2017), this discrepancy was shown to arise from a combination of DFT error and the neglect of vibrational enthalpy. By consideration of the relative phase stability of the np and lp structures, it is seen that the benchmarked MGGA functional actually performs worse for dry MIL-53(Cr) than the ad hoc one. Nonetheless, we use the benchmarked TPSS + D3(BJ) + U + J MGGA functional in this work because of its known transferability across different systems. One must keep in mind that the results of this work will tend to show overbinding of the np structure with respect to the lp one; but at fixed volume, one expects interactions involving water to be reasonably accurate.
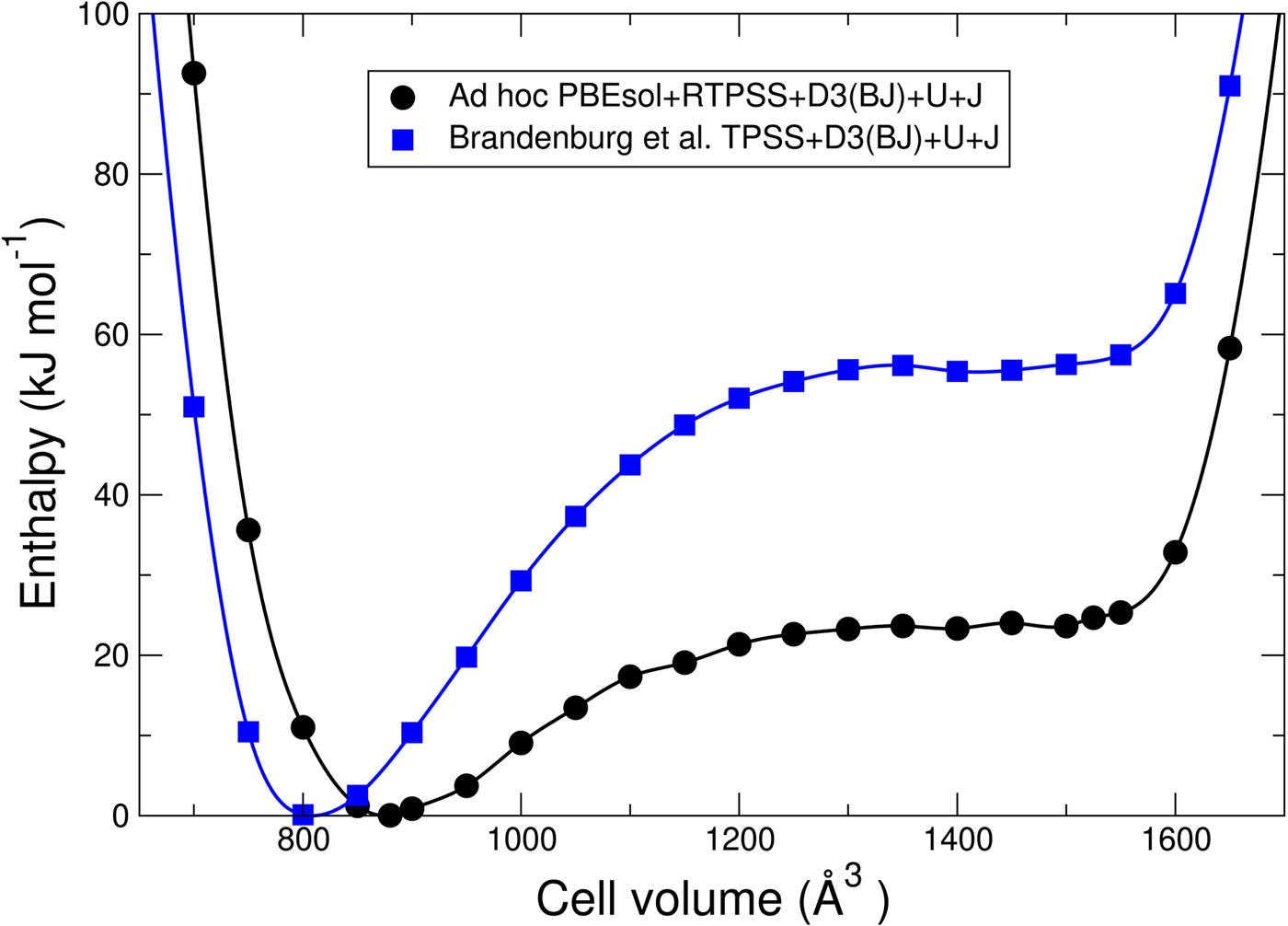
Figure 2. Comparison of enthalpy vs. volume of dry MIL-53(Cr) using an ad hoc parameterization of DFT at the MGGA level (PBEsol + RTPSS + D3(BJ) + U + J) (Cockayne and Nelson, Reference Cockayne and Nelson2015; Cockayne, Reference Cockayne2017) and a MGGA that has been benchmarked across different systems (TPSS + D3(BJ) + U + J) (Brandenburg et al., Reference Brandenburg, Bates, Sun and Perdew2016).
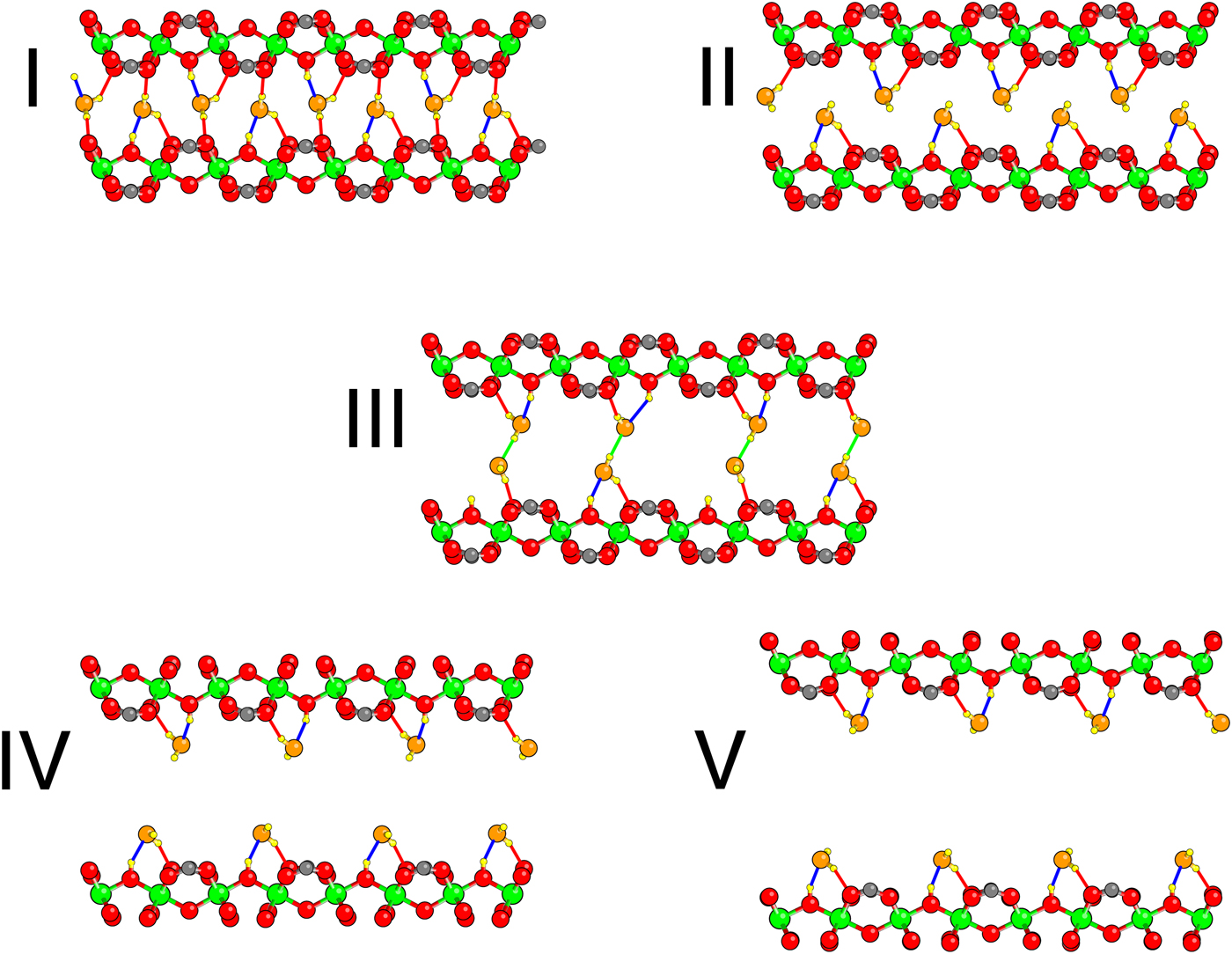
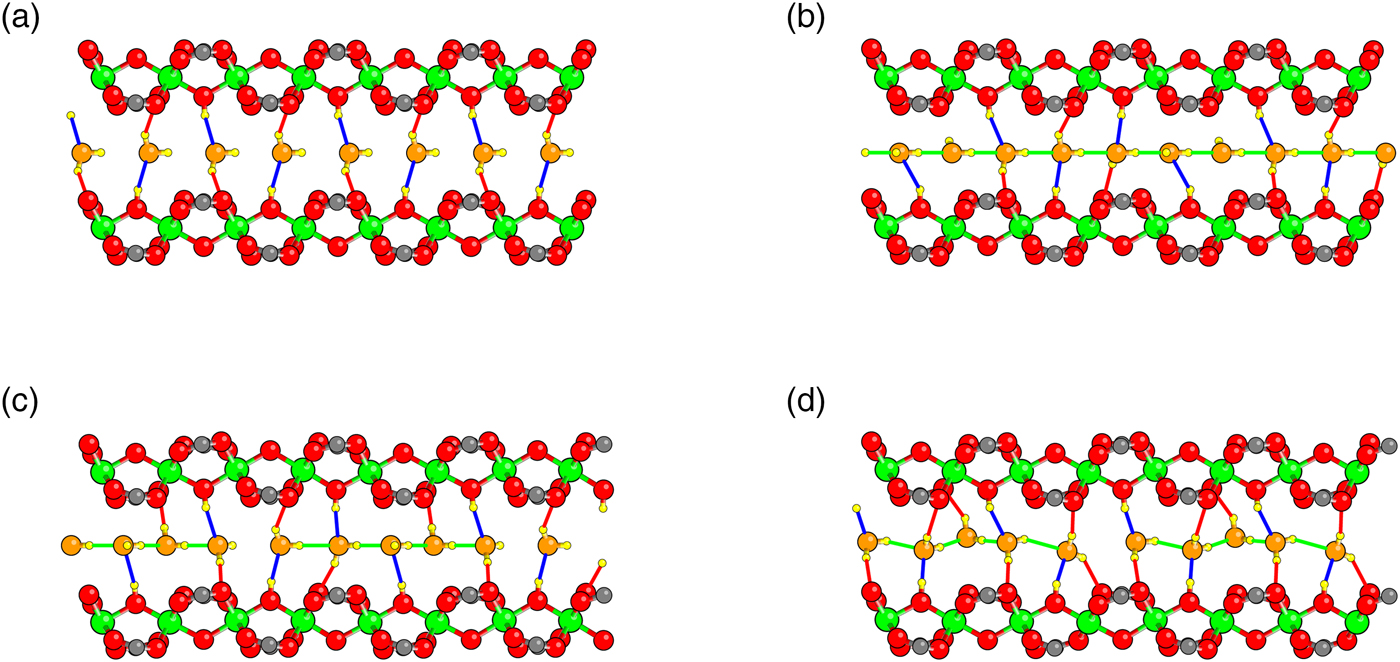
Having established the quality of our density functional on dry MIL-53(Cr), we move to the interaction of MIL-53(Cr) with water. We first present results for composition x = 1.0 H2O per Cr. Structures were initialized both in the lp phase and np phase. Given previous results, one OW was set at a hydrogen bonding distance from the H of each hydroxyl group in the framework; the H positions of the water molecules were initially randomized. The exploration of structure vs. volume was performed as described in the Methods section. In all, five different configurations are found (Figure 3), whose calculated enthalpies as a function of volume are shown in Figure 4.

Figure 3. Five different relaxed configurations found during DFT studies of MIL-53 hydrated at x = 1, in order of increasing cell volume. In contrast to Figure 1, the view is down the a-axis, and the tunnel axis runs left–right. Cr atoms are green, framework O red, OW orange and H yellow. O–H bonds, defined as O and H separated by <2.3 Å, are shown: hydrogen bonds between O W and framework are blue, bonds between H W and framework red, and hydrogen bonds between water molecules green.
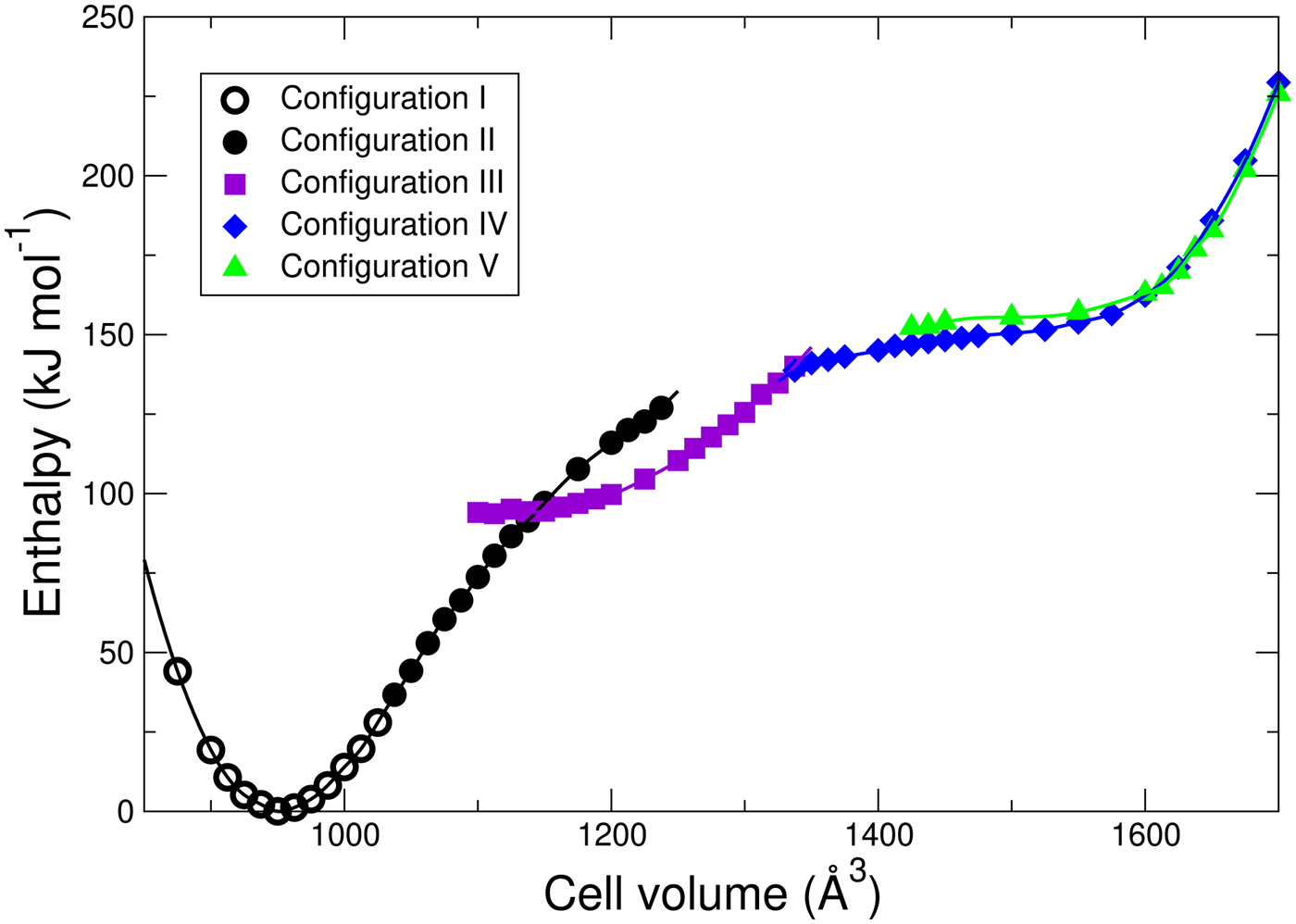
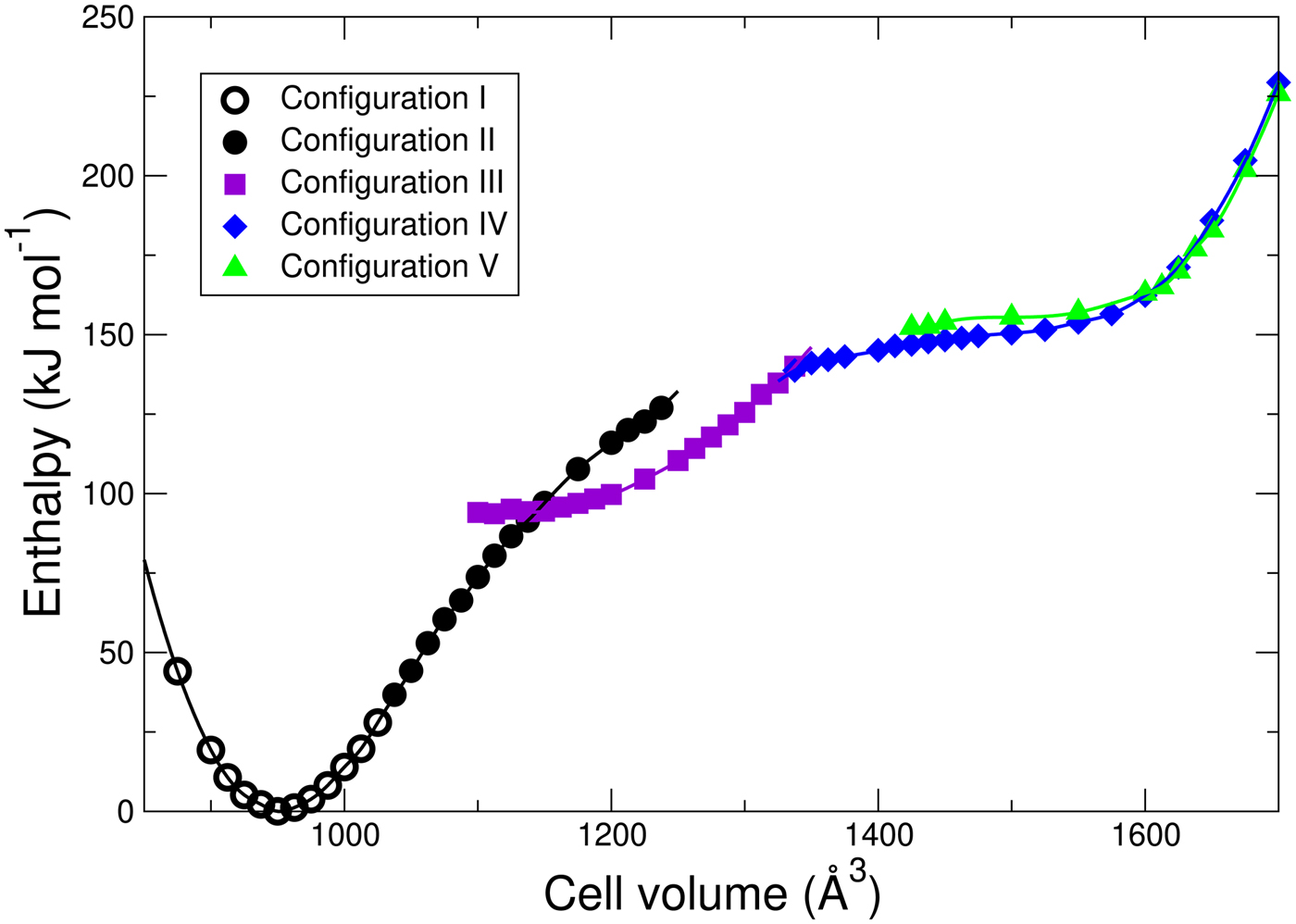
Figure 4. Energetics of H2O adsorption in MIL-53 as a function of volume and configuration type shown in Figure 3.
Four of the five configurations (I, II, IV,V) have all of the OW hydrogen bonded to a framework H and one HW of each H2O forming a bond with a framework O on the same side of the pore. In the np state (configuration I), the pore becomes so narrow that the other HW of each H2O forms a bond with a framework O on the opposite side of the pore. The geometry at intermediate volumes (configuration III) is different. Consecutive pairs of water molecules along the pore dimerize. In doing so, some of the hydrogen bonds between OW and the framework are broken. Inspection of the geometry of the water molecules in this dimer shows that it is very similar to that of an isolated water dimer. Note that the H2O bonding arrangements at the top of the pore differ between configurations II and IV. Configurations IV and V differ subtly as can be observed by differences in the orientations of the HW.
Note that the “dimer” configuration III interrupts what is essentially a smooth energy curve for each configuration with each OW hydrogen bonded to the framework. The transition from configuration I to configuration II is not precisely defined because of continuous stretching of the O–H bonds across the pore, but occurs roughly at a cell volume roughly 1030 Å3. The other transitions are between geometrically distinct configurations and the transition volumes can thus be defined as those at which the enthalpies cross: II–III at 1144 Å3; III–IV at 1331 Å3, and IV–V at 1604 Å3. As the volume decreases, the DFT relaxation of configuration V spontaneously transforms into configuration IV at a cell volume of about 1400 Å3; while upon expansion configuration IV remains metastable until at least cell volume 1725 Å3. The dimer of configuration III spontaneously breaks up into the separate water molecules into configuration IV and vice versa at a volume not far from the equilibrium volume. Configurations II and III remain metastable up to at least the volumes shown in Figure 4.
For x = 0, the calculated enthalpy difference between the np and lp phases is 55 kJ mol−1. For x = 1, the difference is approximately 150 kJ mol−1. While our results incorrectly predict the np phase to be more stable, the experimental free energy of the lp phase of MIL-53(Cr) is approximately 12 kJ mol−1 lower than that of the np phase at room temperature (Coombes et al., Reference Coombes, Corå, Mellot-Draznieks and Bell2009; Coudert et al., Reference Coudert, Jeffroy, Fuchs, Boutin and Mellot-Draznicks2008; Devautour-Vinot et al., Reference Devautour-Vinot, Maurin, Henn, Serre, Devic and Férey2009; Beurroies et al., Reference Beurroies, Boulhout, Llewellyn, Kuchta, Férey, Serre and Denoyel2010). Assuming that the enthalpy difference change of 95 kJ mol−1 from x = 0 to x = 1 is correctly computed by the DFT calculations, that this change is linear in composition, and that changes in free energy can be approximated by changes in enthalpy, we estimate that the transition from the lp phase to the np phase in hydrated MIL-53(Cr) will occur at a composition near x = 0.13.
While Guillou et al. (Reference Guillou, Millange and Walton2011) reported evidence for hydrogen-bound water dimers in np MIL53(Cr), our results showed dimers only at intermediate volumes. We thus performed further explorations of water in the np phase. Note that the average OW–OW separation distance along a tunnel in MIL-53(Cr) is about 3.4 Å, which is not a favorable distance for hydrogen bonding. We note, however, that the doubled MIL-53(Cr) c lattice parameter of about 13.6 Å that we use in this study could accommodate 5 H2O per repeat distance with favorable OW–OW distances for hydrogen bonding. This led us to study the relaxed geometries of np MIL-53(Cr) with x = 1.25 H2O per Cr.
Five H2O molecules were initially placed in the MIL-53(Cr) np structure such that an OW and HW of each H2O were along the central axis of the tunnel with each HW pointing toward the OW of the next molecule [Figure 5(b)]. One water molecule had its other HW in the ac plane. The other water hydrogens were successively rotated by an angle of 2π/5. With this arrangement, not only is there a hydrogen bond between each successive pair of water molecules, but each hydroxyl unit forms a hydrogen bond with one water molecule, and there are some additional bonds between HW and carboxylate oxygens as in the x = 1 case.
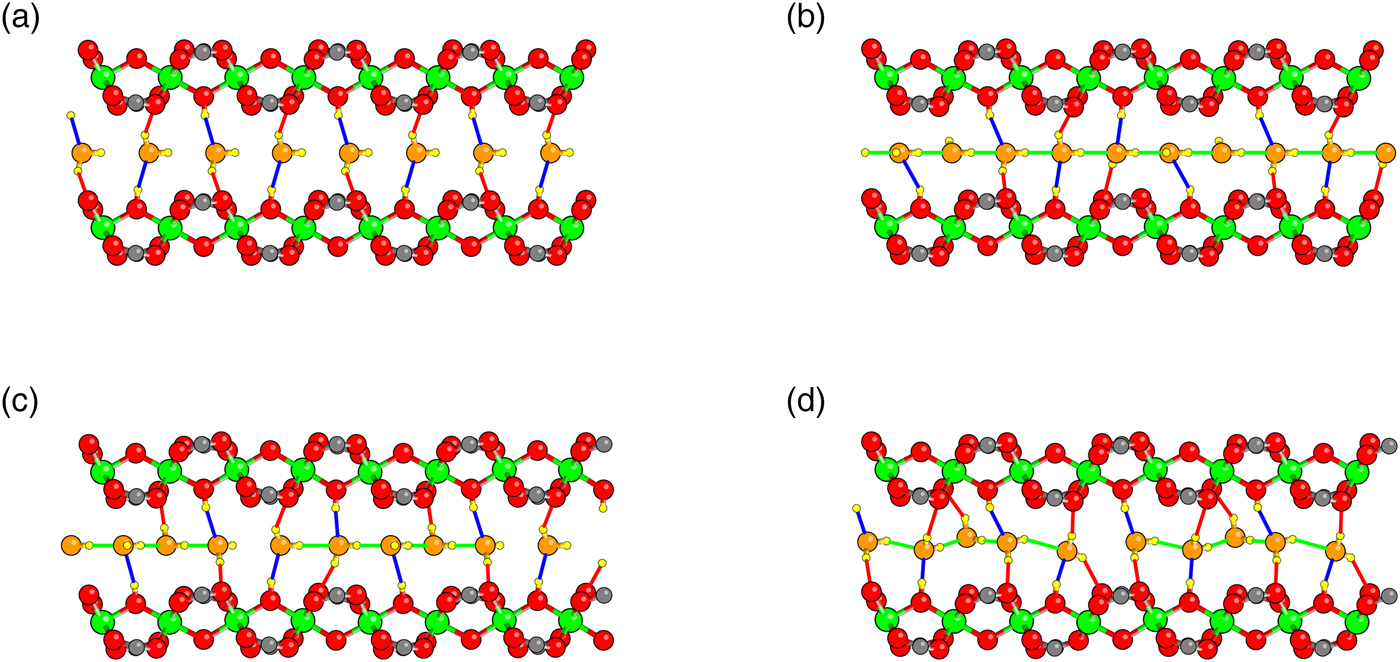
Figure 5. Hydrogen bonding between water molecules in and configuration type. (a) Average separation of 4 H2O per repeat distance is too long for hydrogen bonding. (b) Initialization of structure with 5 H2O per repeat distance. (c) Constrained relaxation. (d) Full relaxation.
We first relax the x = 1.25 structure with the constraint that the OW and HW originally involved in the hydrogen bonding remain along the tunnel axis. The results are shown in Figure 5(c). Note that one interwater hydrogen bond is broken to improve interactions with the framework. We then perform a full relaxation. In the full relaxation [Figure 5(d)], a different hydrogen bond is broken, and water molecules show noticeable displacement from the central axis. Our results predict that the number of intermolecular H bonds in hydrated MIL-53(Cr) should dramatically increase as the composition is raised above x = 1.
IV. DISCUSSION
Throughout the text, we have been careful to use the term “hydrogen bond” to describe bonding between water molecules and binding of a water oxygen OW to the H of the hydroxyl unit of the MIL-53(Cr) framework. On the other hand, we have simply used the term “bond” to describe the interaction between water hydrogens HW and carboxylate oxygens, even though the species involved are the same and separation distances are similar in both cases. We base our terminology on the ab initio molecular dynamics results of Haigis et al. (Reference Haigis, Coudert, Vuilleumier and Boutin2013), where they find that the HW–carboxylate oxygen interactions are not true hydrogen bonds, because (at the 350 K simulation temperature), the interaction of a HW with an individual carboxylate atom had only a short lifetime (typically <200 fs). Our results are essentially zero temperature results ignoring zero-point vibrational motion. Under these conditions, the minimum energy configurations do show HW bonded to carboxylate oxygens at a typical distance of about 1.8 Å to 1.9 Å typical of hydrogen bonding. It would be interesting to calculate the shape and depth of this binding well in comparison with the zero-point vibrational energy of an HW in this well to see if the H is truly bound at zero temperature.
Guillou et al. (Reference Guillou, Millange and Walton2011) used structure refinement results showing a split O W position to conclude that water dimers formed in np MIL-53(Cr). Our results present an alternate interpretation. In configuration I of Figure 3, the H2O with OW bound to the bottom of the pore tilt to the right. To reproduce the parent C2/c symmetry, equivalent configurations with the H2O tilted to the left must be included. Supposing that an actual np structure has a statistical mix of these configurations locally, then the average crystallographic structure would have a split OW position. The difference between the OW position of our symmetrized configuration I with the OW position in the Supplementary Material of Guillou et al. (2011) is only 0.2 Å. We conclude that the split OW position seen in the crystallographic refinement does not necessarily imply that the water molecules form dimers.
The energetics of the global water configurations shown in Figure 3 have interesting implications if they apply to local pore closing dynamics in MIL-53(Cr). The results imply that, as H2O is added to the lp structure of dry MIL-53(Cr), H2O attached to consecutive OH units on opposite sides of the pore could attract each other to form a dimer, first accelerating the pore closing as seen in the energy vs. volume curve of configuration III (Figure 4). However, the energy barrier between configuration III and configuration IV that is implied by the metastability of each of these configurations into the volume domain of the other suggests that complete closing of the pore is somewhat hindered.
V. CONCLUSIONS
The energetics of water adsorption into MIL-53(Cr) was investigated using DFT at the MGGA level. Various types of important O–H interactions were identified. At 1.0 H2O per Cr, for volumes intermediate between those of the np and lp structures, a low-energy configuration was found where all water molecules are paired into dimers. Our results provide a basis for the interpretation of experimental, theoretical and modeling work on water adsorption up to 1.25 H2O per Cr.