Introduction
Genetic diversity forms the evolutionary potential of biological species. It enables natural populations to adapt to changing environmental conditions, and is exploited in plant breeding programmes for the development of novel crop varieties in response to new biotic and abiotic stresses and changing consumer preferences. Genebanks play an important role in safeguarding this genetic diversity for future availability and in providing genetic resources to the research and plant breeding community for short-term use (Engels and Visser, Reference Engels and Visser2003).
Since the 1970s, a wide variety of molecular marker technologies have emerged to study genetic diversity at the DNA level, and these techniques have found their way to many biological disciplines (Agarwal et al., Reference Agarwal, Shrivastava and Padh2008; Mir and Varshney, Reference Mir, Varshney and Henry2012). For example, molecular marker technologies such as restriction fragment length polymorphism, random amplified polymorphic DNA, simple sequence repeat (SSR) and amplified fragment length polymorphism (AFLP) have been widely applied in many crops for the identification of marker–trait associations and the construction of genetic maps in order to gain insight into the genetic basis and genomic organization of traits of interest (Mauricio, Reference Mauricio2001). In modern plant breeding, molecular marker technologies have proven useful to facilitate the breeding process through marker-assisted selection (Collard and Mackill, Reference Collard and Mackill2008). These technologies have also been adopted by the genebank community in order to obtain more detailed information about the genetic structure of genetic resources collections and to support various management procedures thereof (Spooner et al., Reference Spooner, van Treuren and de Vicente2005). Each of the molecular marker technologies has its own characteristics, and each differs from one another with respect to the methodology involved, its ease of use and interpretation and the genomic regions that are targeted in the DNA. Therefore, the choice of the marker system should depend on the specific application of the study (Spooner et al., Reference Spooner, van Treuren and de Vicente2005). Marker technologies do have in common that only a subset of the genome is sampled, while full genome coverage can be reached only through the complete sequencing of an individual's genome.
Especially in the field of sequencing, tremendous technological advances have occurred over the last decade. Due to these developments, the amount of data output has increased tremendously, while at the same time, the production costs of sequence data have decreased dramatically. These novel high-throughput technologies are collectively denoted as next-generation sequencing (NGS). NGS technologies are expected not only to revolutionize genomics research, but also to fundamentally change the management of genetic resources collections and the services provided by genebanks to the user community (Kilian and Graner, Reference Kilian and Graner2012; McCouch et al., Reference McCouch, McNally, Wang and Sackville Hamilton2012). Here we argue that in the short term, NGS technologies will have an impact on the user-oriented activities of genebanks, rather than on their housekeeping operations, while the nature of the changes in genebank activities is expected to be driven by the user community. The main goal of the present study was to explore how the developments in NGS technologies may affect the operations of genebanks and, consequently, how genebank agendas may have to change.
The sequencing era
DNA sequencing dates back to the 1970s when, among others, the chain termination method was introduced by Sanger. Classical Sanger sequencing included the laborious procedures of cloning DNA fragments, radiolabelling, polyacrylamide electrophoresis and manual scoring of autoradiograms. The introduction of automated sequencers in the 1980s greatly facilitated Sanger sequencing through the use of capillary electrophoresis, fluorescent labelling and automated scoring software. In the 1990s, the introduction of microchip technology enabled the identification of small sequence variations in previously established DNA sequences through the hybridization of DNA fragments to large arrays of short synthetic oligonucleotides. A major leap forward in sequencing throughput was provided by the introduction of the 454 technology in 2004 and the Solexa technology in 2006, both capable of analysing millions of DNA fragments simultaneously. Both technologies use the polymerase chain reaction (PCR) to generate large numbers of copies per fragment and cyclical primer extension to synthesize DNA strands that are complementary to single-stranded DNA fragments. Incorporated nucleotides during primer extension are reported by chemiluminescence (pyrosequencing) in the 454 sequencing and by fluorescence in the Solexa sequencing. Advances in detection techniques did no longer require the multiplication of DNA fragments by PCR, which formed the basis of the introduction of the true single-molecule sequencing technology by Helicos BioSciences in 2009, the single-molecule real-time technology by Pacific Biosciences in 2010 and Ion Torrent's semiconductor sequencing by Life Technologies in 2011. A fundamentally different technology to analyse intact single DNA molecules is offered by nanopore sequencing, which was introduced in 2012. This technology uses array chips consisting of thousands of protein nanopores, immersed in a salt solution and exposed to an electric field. Single-stranded DNA molecules are fed through the nanopore, one base at the time, resulting in a characteristic change in the current, depending on the nucleotide passing through the nanopore. As a result of these developments, sequencing throughput of NGS technologies has increased tremendously during the last decade. For example, the Illumina HiSeq 2500 with a dual flow cell system is capable of producing 108 Gb of Solexa sequence reads per 24 h (Illumina, 2013), which is equivalent to 36 times the human genome size and to approximately 67,000 times the throughput of classical Sanger sequencing. At the same time, the production costs, including library construction, labour, utilities, reagents, consumables, administration and instruments, per Mb of analysed DNA sequence plummeted from approximately 5000 USD in 2001 to 7 cents in 2012 (Wetterstrand, Reference Wetterstrand2013), although, to date, sequencing costs are no longer continuously decreasing (Hall, Reference Hall2013). The characteristics of, and the developments in, sequencing technologies have been described in more detail by Egan et al. (Reference Egan, Schlueter and Spooner2012), Thudi et al. (Reference Thudi, Li, Jackson, May and Varshney2012), Ekblom and Galindo (Reference Ekblom and Galindo2011), Delseny et al. (Reference Delseny, Hanb and Hsing2010), Edwards and Batley (Reference Edwards and Batley2010), Metzker (Reference Metzker2010) and Varshney et al. (Reference Varshney, Nayak, May and Jackson2009).
As a result of the technological advances, the complete genomes of individuals of over 180 organisms have been sequenced since 1995 (Ruder and Winstead, Reference Ruder and Winstead2013), including more than 50 (crop) plant species (CoGePedia, 2013). In order to identify genomic variation, initiatives are increasing to sequence multiple individuals from organisms for which a draft or reference sequence has been established previously, such as for maize (Jiao et al., Reference Jiao, Zhao, Ren, Song, Zeng, Guo, Wang, Liu, Chen, Li, Zhang, Xie and Lai2012) and rice (Huang et al., Reference Huang, Lu and Han2013). Resequencing methods are usually directed to the part of the genome in order to target lower copy regions with higher efficiency, such as through restriction site-associated DNA sequencing (Davey and Blaxter, Reference Davey and Blaxter2010) and genotyping-by-sequencing (Elshire et al., Reference Elshire, Glaubitz, Sun, Poland, Kawamoto, Buckler and Mitchell2011), or to target the more functionally relevant regions, such as by RNA sequencing (Wang et al., Reference Wang, Gerstein and Snyder2009) and exome sequencing (Clark et al., Reference Clark, Chen, Lam, Karczewski, Chen, Euskirchen, Butte and Snyder2011). Furthermore, sequencing methods such as chromatin immunoprecipitation sequencing (Robertson et al., Reference Robertson, Hirst, Bainbridge, Bilenky, Zhao, Zeng, Euskirchen, Bernier, Varhol, Delaney, Thiessen, Griffith, He, Marra, Snyder and Jones2007) and bisulphite genomic sequencing (Lister and Ecker, Reference Lister and Ecker2009) have been developed to identify epigenetic variation resulting from DNA–protein interactions and differences in DNA methylation patterns, respectively.
In genomics approaches, sequencing efforts are primarily directed to the identification of genomic loci that affect variations in traits of interest. Relationships between genomic and phenotypic variations are commonly detected through the analysis of progenies from designed mapping populations (e.g. Wallace et al., Reference Wallace, Larsson and Buckler2013) or by studying individuals from unstructured populations using association genetic approaches (Hall et al., Reference Hall, Tegström and Ingvarsson2010; Rafalski, Reference Rafalski2010; Brachi et al., Reference Brachi, Morris and Borevitz2011). To demonstrate its usefulness, the performance of putative quantitative trait loci is commonly tested across different genetic backgrounds and among various environmental conditions prior to exploitation in plant breeding activities, such as the introgression of novel variation for such quantitative traits in elite materials (e.g. Knoll and Ejeta, Reference Knoll and Ejeta2008). Rather than the identification of individual loci associated with a trait of interest, all available marker data are used to predict phenotypic performance in genomic selection approaches, which is considered particularly useful in breeding programmes for more complex traits (Jannink et al., Reference Jannink, Lorenz and Iwata2010; Lorenz et al., Reference Lorenz, Chao, Asoro, Heffner, Hayashi, Iwata, Smith, Sorrells, Jannink and Sparks2011). Although the advances in sequencing technologies can be expected to boost genomics research and crop improvement, unravelling the genetic basis of biological variation will probably remain a challenging task for many traits. Epistatic interactions between genes, genotype–environment interactions and epigenetic variation are among the factors contributing to the complexity of trait expression. Moreover, to relate the wealth of NGS data to trait variation, the generation of high-throughput, high-quality and reliable phenotypic data has become the next bottleneck (Furbank and Tester, Reference Furbank and Tester2011; Cobb et al., Reference Cobb, DeClerck, Greenberg, Clark and McCouch2013), while the technological developments continue to challenge bioinformaticians for innovations regarding the analysis and management of the massive amounts of sequencing data (Horner et al., Reference Horner, Pavesi, Castrignanò, D'Onorio De Meo, Liuni, Sammeth, Picardi and Pesole2010; Lee et al., Reference Lee, Lai, Lorenc, Imelfort, Duran and Edwards2012).
Next-generation genebanking
As NGS technologies enable the efficient sequencing of large numbers of samples, and genebank collections generally consist of many poorly studied and underutilized accessions, it is not surprising that the impact of NGS technologies on the functioning of genebanks has also received attention. It has been suggested that the advances in sequencing may fundamentally change the functioning of genebanks, both in their collection management procedures and in the services provided to the user community (Kilian and Graner, Reference Kilian and Graner2012; McCouch et al., Reference McCouch, McNally, Wang and Sackville Hamilton2012).
These high expectations, however, may not be valid for the genebank community at large. For example, at the research centres of the Consultative Group on International Agricultural Research (CGIAR), the focus is on a single or just a very limited number of staple crops, while ample research facilities and expertise are usually present. Consequently, the CGIAR genebanks are better positioned to profit from genomics research, in contrast to many institutional and national genebanks that manage a variety of crops and often have limited access to facilities and expertise. Even such genebanks may strongly differ in the ability to access modern technologies and to adapt to changing demands, as between national genebanks of developed and those of non-developed countries. Application of NGS technologies is therefore less straightforward for most genebanks when compared with genebanks in institutes such as the International Rice Research Institute and the International Maize and Wheat Improvement Center (CIMMYT). It is therefore not surprising that regarding sequencing applications, most progress is achieved for crops such as rice and maize (McCouch et al., Reference McCouch, McNally, Wang and Sackville Hamilton2012). Considering the funds that can be raised for such important and highly commercial staple crops, it remains to be seen whether similar advances in applying NGS technologies can ever be realized for less important crops.
An additional question is whether NGS technologies can be expected to influence all genebank activities. Regarding collection management, NGS technologies could be useful to basically support all management areas. For example, DNA sequence data of genebank accessions may be used to determine the genetic structure of collections and to improve the composition thereof by eliminating redundancies (Van Treuren et al., Reference Van Treuren, Engels, Hoekstra and van Hintum2009). Ample sequence data of the existing collection allow genebank curators to take more informed decisions about acquisition by evaluating potentially interesting materials for their added value to the genetic diversity already present in the collection (Van Treuren et al., Reference Van Treuren, van Hintum and van de Wiel2008). NGS data could also be used to monitor the regeneration of accessions in order to ensure the maintenance of genetic integrity thereof, for example, by comparing sequence data of samples before and after regeneration (Van Hintum et al., Reference Van Hintum, van de Wiel, Visser, van Treuren and Vosman2007). Considering these applications to collection management, the possibilities of NGS technologies largely resemble those offered by molecular markers, such as SSRs and AFLPs, albeit NGS technologies allow for higher analytical power. With regard to their impact on genetic resources management, molecular markers were once expected to revolutionize the functioning of genebanks (Van Hintum and van Treuren, Reference Van Hintum and van Treuren2002). Although these technologies have proven very useful to address a wide range of questions related to germplasm management (Spooner et al., Reference Spooner, van Treuren and de Vicente2005), their systematic and routine use has so far not been introduced by genebanks. The reasons for this probably include insufficient molecular and bioinformatics expertise and insufficient budget to finance molecular analyses. In other words, as long as NGS technologies do not become trivial in terms of costs and operation, it is unlikely that NGS technologies will be adopted by genebanks for routine use in their collection management, and it is considered unlikely that this situation will change in the short term. However, advances in NGS technologies can rather be expected to have an impact on the user-oriented services of genebanks.
Regarding the new developments, genebanks may choose to follow a strict conservative approach by considering NGS applications as the domain of the user community, and by arguing that the user community is best served by strengthening the core business of genebanks, such as improving the quality of collection management procedures and providing access to a wider diversity of a crop's gene pool. Alternatively, genebanks may decide to keep an open mind for novel approaches by considering NGS applications as a driver for innovation, and by arguing that the user community can be better supported by developing new services, such as novel, user-oriented collection types and information services. Such potential approaches will be discussed in more detail in the sections below, while also the possibilities for cooperation with the user community and the development of strategic alliances will be addressed.
Improving the quality of genebank procedures
Ensuring availability of genetic resources is a key responsibility of genebanks, while distribution of high-quality germplasm samples is essential to link conservation with utilization (Engels and Visser, Reference Engels and Visser2003). Because the probability of identifying genomic variants in resequencing efforts depends on the available variation for the design of diversity panels, optimal access to the germplasm contained in genebanks is required. As such, this can be considered a prerequisite for genebanks to enter the sequencing era. However, many genebanks offer only poor access to their accessions and often distribute material with poor seed viability and/or little documentation.
Genebanks may increase their efficiency and effectiveness by introducing a quality management system. Important elements of a quality management system are the detailed documentation of operating procedures and the independent monitoring to check whether practices are in line with these procedures. In addition, a quality management system is explicitly aimed at the improvement of operating procedures. The issue of quality management procedures is receiving increasing interest from the genebank community. For example, a quality management system has been introduced at the Centre for Genetic Resources, the Netherlands (CGN) and at the German genebank located in the Leibniz-Institut für Pflanzengenetik und Kulturpflanzenforschung in Gatersleben. In the same vein, the Global Crop Diversity Trust is exploring possibilities to monitor the quality of the CGIAR's genebank operations. Recently, a quality management system has been introduced at CIMMYT.
Besides securing operating quality, developments are ongoing to standardize genebank procedures. Genebanks may start working according to agreed operational standards, aimed at the successful conservation and availability of plant genetic resources (PGR; e.g. Thormann et al., Reference Thormann, Yang, Allender, Bas, Campbell, Dulloo, Ebert, Lohwasser, Pandey, Robertson and Spellman2013). Existing genebank standards have recently been revised by the Food and Agriculture Organization of the United Nations (CGRFA, 2012). These standards will form the basis for the development of a quality management system to be used within the European Genebank Integrated System (AEGIS) of the European Cooperative Programme for Plant Genetic Resources (ECPGR), which aims to establish a virtual European genebank collection (Engels and Maggioni, Reference Engels, Maggioni, Maxted, Dulloo, Ford-Lloyd, Frese, Iriondo and Pinheiro de Carvalho2012). The AEGIS Memorandum of Understanding has been signed by 33 countries from the European region (AEGIS, 2013). Adoption of the agreed minimum standards by the associated genebanks will ensure proper management practices, resulting in the availability of high-quality germplasm for distribution to the user community.
Providing access to a wider diversity
In addition to providing optimal access to high-quality information and material, the user community also expects genebanks to develop collections that optimally represent the crop's gene pool. A wider diversity of genebank collections is, for instance, essential to optimally design diversity panels for resequencing projects aiming at the identification of novel genomic variants. Therefore, a proper representation of the gene pool of a crop can be considered another prerequisite for genebanks to move into the sequencing era. However, current genetic resources collections, either individually or collectively, often show a rather poor representation, and hence should be optimized (Van Treuren et al., Reference Van Treuren, Engels, Hoekstra and van Hintum2009). Especially, crop wild relatives are regarded as an increasingly important asset of genetic resources collections, but, nevertheless, they are under-represented in genebanks (Maxted et al., Reference Maxted, Kell, Ford-Lloyd, Dulloo and Toledo2012). More detailed insight is needed into the structure of a crop's gene pool, the importance of the involved species for utilization, and the gaps that exist in current collections in order to guide acquisition strategies by prioritizing species and distribution areas for collecting expeditions (Van Treuren et al., Reference Van Treuren, Coquin and Lohwasser2012). Geographic information systems are increasingly being used to predict species occurrence in thus far unexplored areas, while integration of the distribution models with climate change scenarios enables the prediction of species or areas expected to be threatened in the future (Jarvis et al., Reference Jarvis, Lane and Hijmans2008; Maxted et al., Reference Maxted, Dulloo, Ford-Lloyd, Iriondo and Jarvis2008; Ramírez-Villegas et al., Reference Ramírez-Villegas, Khoury, Jarvis, Debouck and Guarino2010). This strategy to ensure conservation and future availability of wild relatives of a wide range of crops listed on Annex 1 of the International Treaty is currently followed in the project entitled ‘Adapting agriculture to climate change: collecting, protecting and preparing crop wild relatives’, which is coordinated by the Global Crop Diversity Trust (Crop Wild Relatives and Climate Change, 2013). Obviously, collecting missions may also be organized under the flag of the ECPGR or by individual genebanks.
Introducing novel collection types
A potential new service of genebanks is to develop novel collection types to better address the needs of specific user groups, such as the genomics research community. Currently, genebanks maintain a single collection for conservation and use, but, in fact, accessions may be unsuitable to serve both purposes at the same time. In this context, the CGN has started to receive requests from publicly funded genomics projects, such as the ‘150 Tomato Genome ReSequencing Project’ (WUR, 2013) and the ‘Compositae Genome Project’ (CGP, 2013) to store research material and to make it available to the wider user community. It is important to note that this material usually does not serve a conservation purpose, as it involves selected lines from already existing genebank accessions or mapping populations with limited diversity. This issue may be addressed by introducing different collection types for different purposes. Evidently, the prime type of collection for a genebank should be the conservation collection, which is, or should be, the type of collection that genebanks currently maintain. The conservation collection primarily serves a long-term conservation goal, while it also is accessible to the user community. However, in addition, specific user-oriented collection types may be developed, such as those consisting of populations that can be offered as sets of lines. Such ‘special sets’ may include mapping populations such as recombinant inbred lines, near-isogenic lines and multi-parent advanced generation inter-crosses. Another collection type may consist of genetically purified lines that, for example, were used in resequencing experiments for allele mining purposes. In order to unambiguously relate phenotypic observations to genotypic data, purified seed stocks rather than heterogeneous genebank accessions are required. For instance, it has been suggested to develop core reference sets, defined as ‘a set of genetic stocks that are representative of the genetic resources of the crop and are used by the scientific community as a reference for an integrated characterization of its biological diversity’. Such materials should have been genetically purified, roughly phenotyped and be made publicly, quickly and cheaply available (Glaszmann et al., Reference Glaszmann, Kilian, Upadhyaya and Varshney2010), and could be used to gradually build up ‘allele collections’. Considering their importance for the genomics research community and the fact that numerous genetic stocks are already present at institutes without optimal conservation facilities, collecting and distributing such materials may form a new responsibility for genebanks and a first priority in developing user-oriented collections. Moreover, genebanks may play a central role in associating the accumulating data with the specific genetic stock.
By distinguishing conservation and specific user activities, the development of several additional collection types could be considered depending on the specific user group. For example, national heritage collections may be developed that regarding documentation, composition and visibility could be tailored to the general public. Another collection type could consist of crop wild relatives that are readily made accessible to the breeding industry but are unthreatened in their natural habitats, and hence do not require conservation.
The consequence of introducing multiple collection types is that they can be subjected to different management regimes. Obviously, the management of the conservation collection should remain aimed at conservation in perpetuity, and material should be accessible and distributed according to the rules and procedures of the International Treaty on Plant Genetic Resources for Food and Agriculture (ITPGRFA) and related instruments. However, the management of the user-oriented collections can be adjusted to the specific user group and the relevance of the material. For example, material for which the usefulness will not last forever, such as probably mapping populations, may be subjected to a low management regime. Moreover, given their interest in specific materials, the user community may be actively involved in certain management operations, such as regeneration. Compared with the conservation collection, material from other collection types could be distributed under alternative conditions, with tailor-made material transfer agreements and/or on the basis of cost recovery, as the ITPGRFA would consider many of the special collections ‘materials under development’ rather than regular genebank accessions. Furthermore, to avoid a potential burden to genebanks, providing sufficient numbers of viable and healthy seeds could be made a prerequisite for the acceptation of special materials by genebanks, while project proposals in which special materials will be generated could budget the initial costs for their conservation.
Developing novel information services
In addition to proper access to biological material, the user community also needs proper access to the relevant information. Again, in this respect, genebank services generally need improvement as accessions are often poorly documented, and phenotypic and genotypic data are poorly, or not, available or accessible, while only limited online search and ordering facilities are present. This situation needs urgent improvement as the value of collections, and thus PGR programmes, depends to a large extent on such information and services.
In this light, it may seem awkward to consider the new potential for genebank information services that can be expected to arise in the sequencing era. Genebanks will be able to tap the genomic information about their resources, and allow the user to search for specific alleles or specific sequences. It will become possible to request a set of accessions with all available variations in a specific allele or sequence, allowing the user to screen a very limited set of accessions knowing that all known allelic variants are included. These new services will depend on the availability of genomic data, i.e. DNA sequences and annotations, and on the automated access to the information. It is considered a matter of time before genebank collections are sequenced and annotated, and genebank and genomic databases are machine readable via the Internet using semantic web or other already available technology.
Even when disregarding the developments in genomics, the possibilities offered by the information and communication technology are currently not used optimally. Information is generally offered by genebanks from a provider's point of view, rather than from a user's point of view. Information services may be improved by developing user-oriented PGR portals, which may serve as an entry point for specific PGR user groups. A portal could be developed for users of lettuce germplasm, for malting barley breeders or for people interested in traditional cultivars suitable for their backyard. These portals should provide access to all information relevant to the specific user group, such as information about the main genebanks from which material can be ordered, where and how user-oriented collections can be accessed, germination and cultivation procedures for materials that are difficult to grow, give links to available phenotypic data and links to the available genomic resources. In addition, the portal may inform users about ordering procedures, including whether material can be ordered online, and if not, who needs to be contacted and how, what kind of material transfer agreement needs to be signed, and whether phytosanitary regulations do apply. The portals can only evolve according to the needs of the user community, and hence they should develop into interactive platforms, where social media may be used to share experiences and ideas. Prototypes of such crop portals have been developed for lettuce (pgrportal.nl/lettuce) and potato (pgrportal.nl/potato) by CGN. However, capacity building can be sought from the ECPGR crop working groups that may adopt the development and maintenance of crop portals as a newly shared responsibility.
Strengthening cooperation with the user community
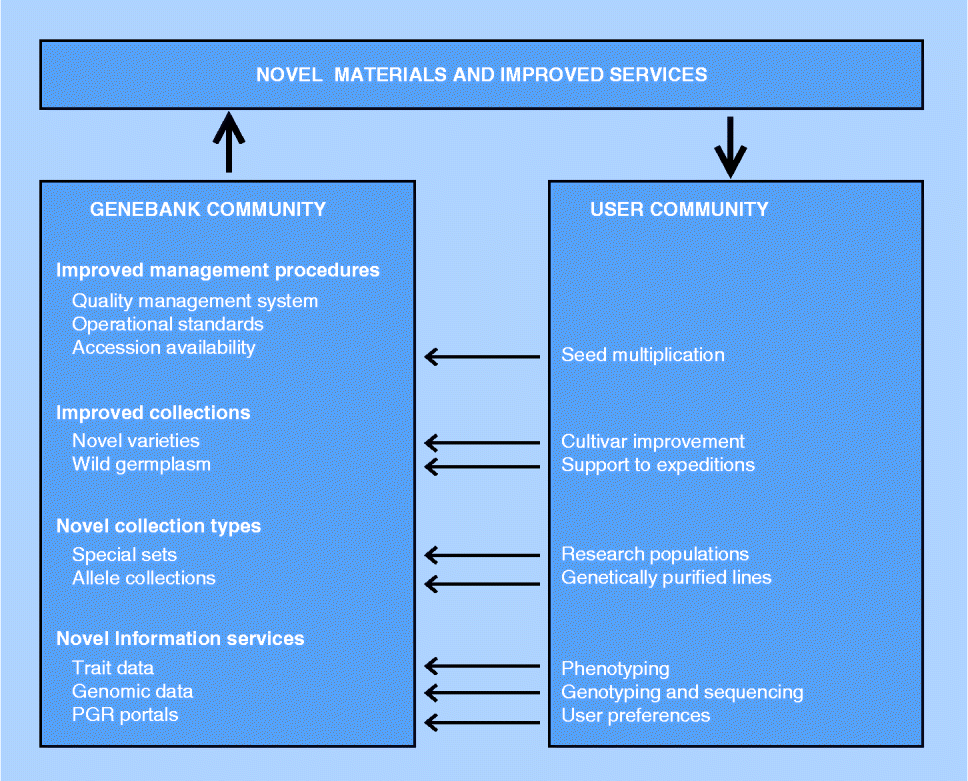
Close cooperation between genebanks and the user community will be a condition for the effectiveness of the user-oriented genebank services. Such cooperation may involve various activities (Fig. 1). Regeneration backlogs at genebanks have been identified as one of the main causes underlying poor collection accessibility, not only preventing utilization of germplasm but also endangering its ex situ survival (Khoury et al., Reference Khoury, Laliberté and Guarino2010). Therefore, cooperation between genebanks and the user community in the regeneration of genebank accessions is of interest to both parties.
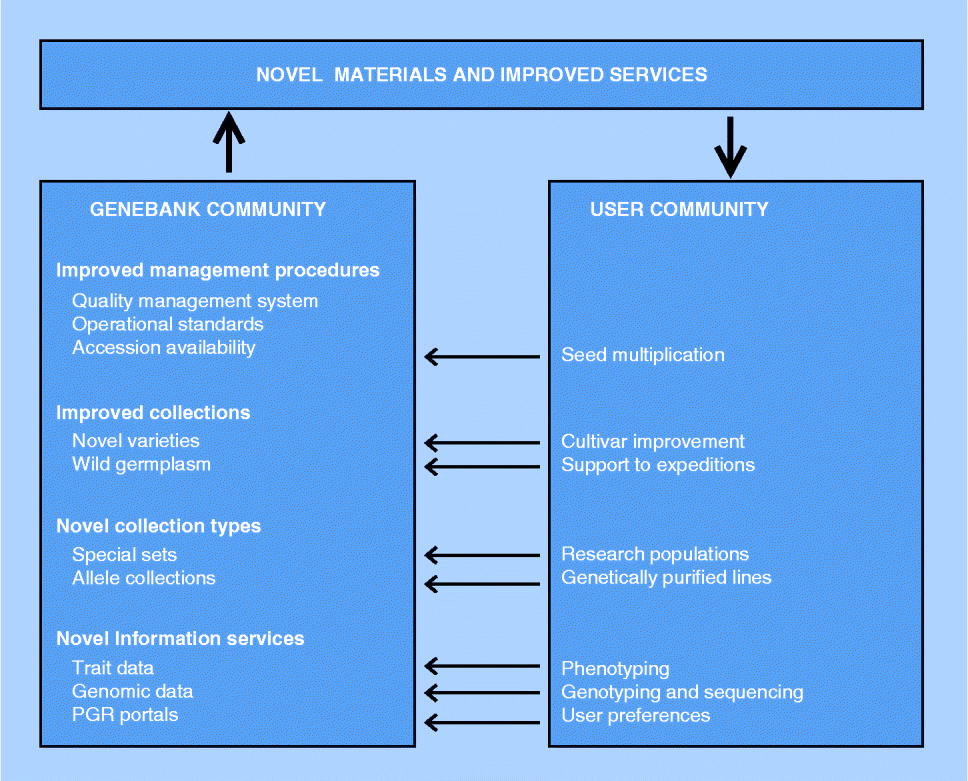
Fig. 1 Elements of next-generation genebanking. Interactions with the user community are indicated by arrows.
Genebanks and users also share an interest in the proper representation of the diversity of a crop's gene pool in collections, which may be ensured through the periodical uptake of commercial varieties with new characteristics (Van Treuren et al., Reference Van Treuren, van Hintum and van de Wiel2008) or by the joint organization of collection expeditions (CGN, 2013). The consequence of an active acquisition programme is that collections may grow beyond a manageable size. In such cases, also the removal of accessions that contribute least to the genetic diversity of the collection may have to be considered (Van Treuren et al., Reference Van Treuren, Engels, Hoekstra and van Hintum2009).
Furthermore, in developing novel collection types, genebanks will have to collaborate with the user community. Choices have to be made regarding the material to be included in the collection as its value will strongly depend on use, and thus on the interest of the user. Moreover, regarding the development of genomics-oriented collections, genetic stocks and mapping populations have to be provided by the user community as genebanks generally neither have the capacity nor have the expertise to produce such materials. Also, in this case, the user community has an interest in cooperation as maintenance of research materials and availability thereof to the wider community is nowadays often required in publicly funded research projects.
Access to phenotypic data allows users to make more well-informed decisions about which materials to order from a genebank, thereby reducing the number of accessions that need to be ordered and processed. The genebank benefits from the more targeted, and hence smaller, requests through reduced seed handling procedures, while seed depletion is slowed down and consequently regeneration frequencies are reduced. However, the availability of phenotypic data is currently rather limited as, due to the high costs involved and the need for specialized research facilities, genebanks generally rely on the user community to provide such data about accessions. In many cases, users are supposed to make their non-confidential experimental results available to the genebank from which the material was ordered, as, for example, requested in the Standard Material Transfer Agreement, but it usually requires substantial efforts from genebanks to obtain such data, if successful at all. In addition, genebanks may proactively organize evaluation programmes in close cooperation with the user community (e.g. Van Treuren et al., Reference Van Treuren, van der Arend and Schut2013). The efficient use of phenotypic data is not only hampered by the lack of availability but also by difficulties in accessibility. Phenotypic data often depend on the applied methodology and experimental conditions, which makes it difficult to present the results in a straightforward manner, let alone that data can be easily searched online. As a consequence, most genebanks are struggling with the presentation of phenotypic data, or have decided not to disclose the information. Standardization of experimental procedures and scoring protocols and the development of ontologies may contribute to the harmonization of phenotypic data, and hence to the improvement of data accessibility (Avraham et al., Reference Avraham, Tung, Ilic, Jaiswal, Kellogg, McCouch, Pujar, Reiser, Rhee, Sachs, Schaeffer, Stein, Stevens, Vincent, Zapata and Ware2008; Shrestha et al., Reference Shrestha, Matteis, Skofic, Portugal, McLaren, Hyman and Arnaud2012). The international aggregated databases such as GENESYS and EURISCO could play an important role in improving data access by developing into firm data repositories that subsequently can be used to feed the PGR portals to be developed with phenotypic data. The PGR portals may play an important role as a central entry point for users looking for specific information or germplasm. However, the development of PGR portals according to user preferences requires a strong interaction between the genebank and user communities.
Strengthening the cooperation with the user community is of particular interest to genebanks that have limited access to research facilities and expertise. Regarding the conservation and utilization of genetic resources, the shared interests of the genebank and user communities may form the basis to agree on common objectives. As such, the cooperation may develop into strategic alliances, in which the involved parties contribute to achieving the agreed goals. Options to develop such strategic alliances not only specifically apply to countries with a strong user community, but may also be organized at the transnational level. In addition, genebanks may seek participation in functional networks providing specific services, such as the European Plant Phenotyping Network (www.plant-phenotyping-network.eu) offering transnational access to infrastructure for the phenotyping of specific traits. In the absence of adequate bioinformatics capacity, collaboration is needed with specialized institutions capable of introducing technologies at genebanks that can handle a wealth of new information and present it in a user-friendly way.
Conclusions
The revolutionizing advances in the field of sequencing and the possibilities thereof for genomics research will not pass the genebank community unnoticed. However, it can be expected that in the short term, the impact will not be so much on the housekeeping procedures of genebanks, but rather on the nature of its services to the user community. Apart from the traditional conservation tasks, genebanks need to optimize their user-oriented services by expanding the range and quality thereof. In this context, strengthening the collaboration with the user community is indispensable.
Acknowledgements
This study was part of the Fundamental Research Programme on Sustainable Agriculture (KB-12-005.03-003) funded by the Dutch Ministry of Economic Affairs. The authors are grateful to Jan Engels, Bert Visser, Sarah Ayling and an anonymous reviewer for their constructive comments on an earlier version of the manuscript.