Introduction
Single-nucleotide polymorphisms (SNPs) and simple sequence repeats (SSRs) are valuable markers for studies on genetic variations existing between diverse genotypes. They are used for quantitative trait loci (QTL) mapping and other genomic applications (Liu et al., Reference Liu, Li, Wu, Chen and Lei2013). The rapid identification of these markers associated with complex, economically important traits in crops has been hindered for most crops by the lack of whole-genome sequence, high-resolution maps and cost-effective platforms for high-density genotyping. Despite numerous genetic marker identification studies being carried out in the recent past, the marker density is not enough to target candidate genes underlying a QTL region and conduct association mapping for complex traits in pepper (Ashrafi et al., Reference Ashrafi, Hill, Stoffel, Kozik, Yao, Chin-Wo and Deynze2012) due to the unavailability of whole-genome sequence (Lu et al., Reference Lu, Cho and Park2012). Limited molecular markers covering the whole genome stand as the major limitation for the development of high-throughput genotyping assays and exploitation of genomic resources for gene discovery and molecular breeding. Large number of markers and cost-effective genotyping technology are both needed for the whole-genome association studies in pepper. It has been demonstrated that high-throughput sequencing of complexity-reduced genomes for marker discovery is a more efficient and effective method for the identification of large numbers of markers (Van Tassell et al., Reference Van Tassell, Smith, Matukumalli, Taylor, Schnabel, Lawley, Haudenschild, Moore, Warren and Sonstegard2008). Next-generation sequencing allows for low-cost genotyping by sequencing, which is useful for discovering and for genotyping of SNPs in various crop species and populations (Spindel et al., Reference Spindel, Wright, Chen, Cobb, Gage, Harrington, Lorieux, Ahmadi and McCouch2013). Thus, the transcriptome assembly of pepper is a major requirement to generate high-quality gene-based molecular markers that are an important resource for the determination of functional genetic variations (Liu et al., Reference Liu, Li, Wu, Chen and Lei2013) and could be used in breeding programmes (Hyten et al., Reference Hyten, Cannon, Song, Weeks, Fickus, Shoemaker, Specht, Farmer, May and Cregan2010). Aiming at the cost-effective identification of putative markers at a low sequencing depth for fine-mapping several QTL regions, herein we report transcriptome profiling and marker discovery from four varieties of pepper, Capsicum annuum, using 454 pyrosequencing technology.
Materials and methods
Plant material and complementary DNA (cDNA) library construction
For RNA extraction using the RNeasy Plant Mini Kit (Qiagen, Valencia, CA, USA) based on the TRIzol RNA isolation protocol, 100 mg of tissues from mature fruits of four pepper (C. annuum L.) varieties (Saengryeg 211, Saengryeg 213, Mandarin and Blackcluster) stored at − 80°C were used. The PolyATract mRNA Isolation System IV (Promega, Madison, WI, USA) was used for purifying mRNA, and the purified products were used to synthesize full-length cDNA using the ZAP cDNA Synthesis Kit (Stratagene, Santa Clara, CA, USA). The Agilent 2100 BioAnalyzer (Agilent Technologies, Deutschland GmbH, Waldbronn, Germany) was used to fragment cDNA for construction of the sequencing library.
454 Pyrosequencing
Library preparation and high-throughput sequencing were carried out according to the manufacturer's instructions (Genome Sequencer FLX Titanium General Library Preparation Kit/emPCR kit/sequencing kit; 454 Life Sciences, Roche Diagnostics, USA, http://www.roche.com) using approximately 1 μg of the adaptor-ligated cDNA population sheared by nebulization.
Sequence processing and assembly and molecular marker discovery
The raw data were deposited in the EBI Sequence Read Archive under the accession numbers Study_IDs ERP001874 Saengryeg 211, ERP001873 Saengryeg 213, ERP001872 Mandarin and ERP001875 Blackcluster. Trimming of low-quality, low-complexity [poly (A)] adaptor sequences and singleton reads was done using the SeqClean version1.0, Lucy program version 2.19. and UniVec build 7.0 software. CLC Assembly cell version 4.6.1 (CLC bio A/S, Aarhus, Denmark) was used for the de novo assembly and identification of all contigs and singletons employing the de Bruijn graph-based approach. Annotation of the assembled reads was performed against the NCBI NR database using the BLAST software with a cut-off e-value of 1.0 × 10− 3. All the isotigs and singletons from transcriptome data were used to mine the SNP, SSR and InDel markers detected by the alignment of individual reads against contigs from the assembly using CLC Genomics Workbench version 4.6.1 and SSRIT (Simple Sequence Repeat Identification) http://www.gramene.org/db/markers/ssrtool (Temnykh et al., Reference Temnykh, DeClerck, Lukashova, Lipovich, Cartinhour and McCouch2001).
Results
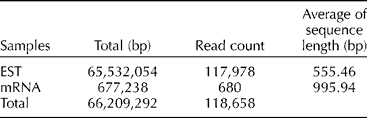
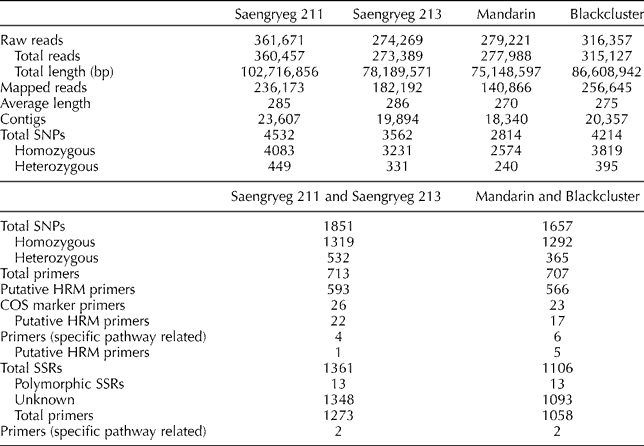
Expressed sequence tags (ESTs) and mRNA sequences used as reference data for the comparison of sequences from the four pepper varieties and the public transcript were collected from the NCBI database (Table 1) (http://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?mode = Info&id = 4072&lvl = 3&lin = f&keep = 1&srchmode = 1&unlock). Transcript fragments (400–600 bp) were produced from cDNA for pyrosequencing. The raw reads generated were filtered for adaptors, primers and low-quality sequences and reduced to high-quality sequences, which included fully assembled reads into contigs, non-overlapping reads and the reads from repeat regions (Table 2). Sequence-based alignments against ESTs submitted to the NCBI database were used to validate the assembled sequences from pooled reads. The reference assembly statistics for molecular marker detection included homozygous and heterozygous genotype SNPs (substitution, insertion and deletion variations) and polymorphic SSRs. To increase the reliability of SNP identification, the results were filtered based on multiple criteria including read depth and allele frequency. The resulting dataset included SNPs distributed across different isogroups of all the four varieties. Based on these, primers for all homozygous SNPs, conserved orthologue set markers and specific pathway-related genes were designed for both isogroups (Saengryeg 211, Saengryeg 213 and Mandarin, Blackcluster) and the putative primers for high-resolution melt analysis were screened. Regarding SSRs, trinucleotide was the most common repeat unit followed by the di-, hexa- and pentanucleotide repeats. Mononucleotide SSRs were excluded because of the frequent homopolymer errors found in the 454 pyrosequencing data. Furthermore, primers for SSR markers and specific pathway-related genes were designed for both the groups. Summary statistics of the identified genetic markers are given in Table 2.
Table 1 Datasets used to analyse mapping of the mRNA sequences of two pepper varieties and the expressed sequence tags (ESTs) and mRNA sequence data of Capsicum annuum in the NCBI database
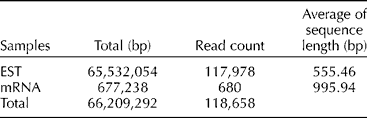
Table 2 Comparative analysis of transcriptome assembly for four pepper varieties (Saengryeg 211, Saengryeg 213, Mandarin and Blackcluster) and single-nucleotide polymorphism (SNP) and simple sequence repeat (SSR) marker and primer details for the two pepper groups (Saengryeg 211 and Saengryeg 213; Mandarin and Blackcluster)
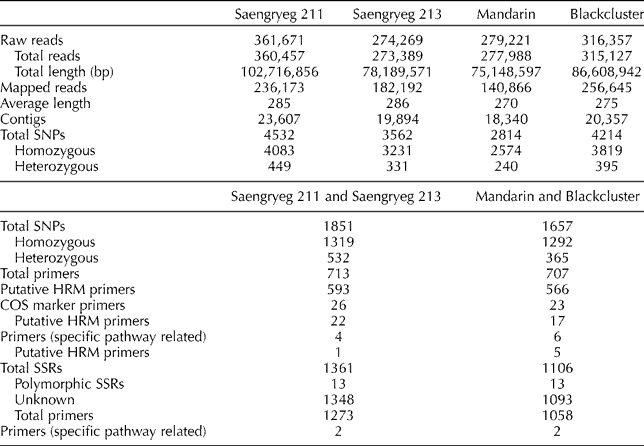
HRM, high-resolution melt; COS, conserved orthologue set.
Discussion
Next-generation sequencing technique has been recently employed in pepper to rapidly generate a large amount of sequence data (Hill et al., Reference Hill, Ashrafi, Chin-Wo, Yao, Stoffel, Truco, Kozik, Michelmore and Van Deynze2013). In the present study, 454 pyrosequencing of four pepper varieties was carried out, where the majority of pepper EST sequences first assembled by Kim et al. (Reference Kim, Baek, Lee, Kim, Lee, Cho, Kim, Choi and Hur2008) were used as reference for the de novo assembly for transcriptome analysis and discovery of numerous markers. The assembly created contigs with larger average lengths than in previously reported systems (Novaes et al., Reference Novaes, Drost, Farmerie, Pappas, Grattapaglia, Sederoff and Kirst2008). The transcriptome assembly of two pepper parental lines (CM334 and Taean) and their hybrid line (TF68) carried out by Lu et al. (Reference Lu, Cho and Park2012) was in accordance with our findings. The present study provides information on numerous molecular markers to be used in breeding programmes in pepper. This high-throughput technique has earlier been reported as an attractive genotyping strategy for SNP discovery for samples characterized de novo (Deschamps et al., Reference Deschamps, Llaca and May2012; Ahn et al., Reference Ahn, Tripathi, Kim, Cho, Lee, Kim, Woo and Cho2014). For the SSRs, the trinucleotide repeats were found to be the maximum, which are more frequently detected in coding regions (Yu et al., Reference Yu, Won, Jun, Lim and Kwak2011). These repeats are generally more robust as they are reported to give fewer ‘stutter bands’ than dinucleotide repeats, and these trinucleotide repeats, in particular, have been demonstrated to be highly polymorphic and stably inherited (Yang et al., Reference Yang, Bao, Ford, Jia, Guan, He, Sun, Jiang, Hao, Zhang and Zong2012). A similar trend has been observed in other species (Sonah et al., Reference Sonah, Deshmukh, Sharma, Singh, Gupta, Gacche, Rana, Singh and Sharma2011). Based on the variety of molecular markers identified in this study, the future marker optimization may be best focused on these after validation. Detection of mutants by comparative marker analysis in these pepper varieties might provide new information on structural and/or regulatory genes that participate in various biosynthetic pathways, thus laying the basis for better understanding the metabolic pathways in chilli pepper.
Acknowledgements
This study was supported by a grant from the Next-Generation BioGreen 21 Program (Plant Molecular Breeding Center No. PJ008024), Rural Development Administration, Republic of Korea.