Introduction
Shortawn foxtail (Alopecurus aequalis Sobol.) is an annual invasive plant of the Poaceae family widespread in North America, Europe and temperate Asia (Zhao et al., Reference Zhao, Li, Guo, Zhang, Ge and Wang2018). A. aequalis (C3-photosynthesis type) is highly adapted to wheat (Triticum aestivum L.; C3-photosynthesis type) planting environments, where its proliferation seriously threatens T. aestivum production worldwide (He et al., Reference He, Gao, Guo and Guo2016). Understanding the genetic diversity of agricultural pests, such as A. aequalis, is important to resolve questions related to their behavioural traits and physiological or genetic constraints and is critical for developing a forecasting system for integrated pest management plans (Sun et al., Reference Sun, Wang, Zhang, Chapuis, Jiang, Hu, Yang, Ge, Xue and Hong2015).
In recent years, molecular markers, such as simple sequence repeat (SSR), have emerged as powerful tools for determining genetic diversity due to their highly distinguishable nature and relatively low cost (Deng et al., Reference Deng, Wang, Zhu, Wen and Yang2015). However, no SSR markers have been developed for A. aequalis due to the laborious and costly collection of expressed sequence tags (ESTs) (Bouck and Vision, Reference Bouck and Vision2007). The lack of SSR markers seriously hampered both basic and applied genomics research in A. aequalis. In our previous study (Zhao et al., Reference Zhao, Li, Bai, Guo, Yuan, Wang, Liu and Wang2017), high-throughput transcriptome sequencing generates a large number of sequences and provides a potential resource for the development of numerous SSRs in A. aequalis. Therefore, this study aimed to (1) develop a comprehensive set of EST-SSR markers based on previous Illumina HiSeq™ sequencing data generated from young leaves and (2) demonstrate their utility using 160 individuals from 20 natural populations of A. aequalis.
Experimental
A total of 20 natural populations of A. aequalis were collected and used in this study (online Supplementary Table S1). Eight individuals from each of 20 natural populations were planted and thus a total of 160 individuals were included. Genomic DNA was extracted from fresh leaves of each individual using the classical cetyltrimethylammonium bromide method with modifications (Porebski et al., Reference Porebski, Bailey and Baum1997).
Genic SSR loci were identified from the unigene set obtained from the A. aequalis transcriptome data by using MIcroSAtellite (MISA, http://pgrc.ipk-gatersleben.de/misa/) identification tool, which is based on the Perl language (Xie et al., Reference Xie, Xiao, Wang, Fang, Liu, Li, Liu, Zhang, Li and Lin2014; Zhao et al., Reference Zhao, Li, Bai, Guo, Yuan, Wang, Liu and Wang2017). Repeat thresholds were adjusted for identification of perfect di-, tri- and tetra-nucleotide motifs with a minimum of 9, 6 and 5 repeats, respectively. Primer pairs in the flanking regions of SSRs for polymerase chain reaction (PCR) were designed with ePrimer3 on-line software (http://www.hpa-bioinfotools.org.uk/pise/eprimer3.html).
PCR amplifications were carried out in 15-μl reaction volumes containing 100 ng of template genomic DNA, 7.5 µl of 2 × Es Taq MasterMix (CWBIO, Beijing, China), 0.6 µl of each primer (10 µM) and variable volume of ddH2O. Amplification products were then separated by denaturing polyacrylamide gel electrophoresis (T-REX, Thermo Scientific, Rochester, USA) and quantified by using a 50 bp DNA ladder (Generay Biotech, Shanghai, China). The number of alleles (Na), effective number of alleles (Ne), observed heterozygosity (Ho), expected heterozygotes (He) and Shannon's information index (I) for each locus were calculated with Popgene software v.1.32 (Yeh, Reference Yeh1997). The polymorphic information content (PIC) for each locus was calculated by an on-line calculator (https://www.liverpool.ac.uk/~kempsj/pic.html).
Discussion
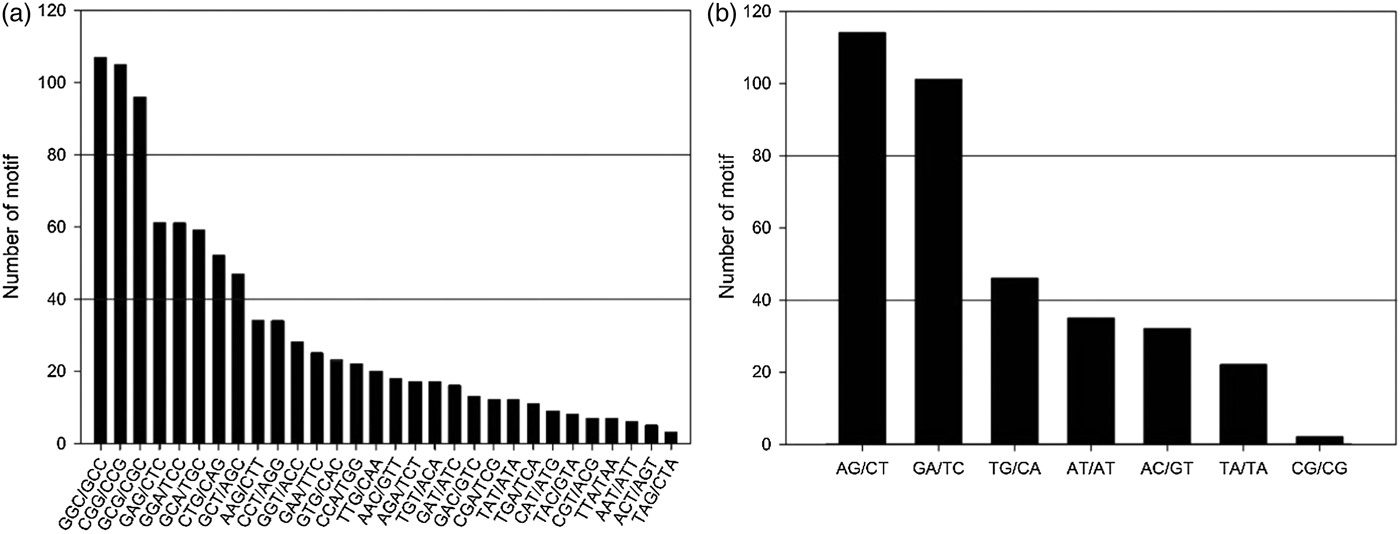
Using the MISA identification tool, a total of 1411 SSR loci were identified from 95,479 unigenes. On an average there was one SSR locus for every 63.9 kb of unigene sequence, corresponding to one SSR for every 67.7 unigenes. In this study, the tri-nucleotide motif (66.27%) repeat motifs was the most abundant type (online Supplementary Fig. S1). The abundant tri-nucleotide motifs in gene regions could elicit the avoidance of frameshift mutation introduction and changes at the protein level (Passos et al., Reference Passos, de Cruz, Emediato, de Teixeira, Azevedo, Brasileiro, Amorim, Ferreira, Martins and Togawa2013). The GGC/GCC repeat was the most abundant tri-nucleotide repeat type (107, 11.4%; Fig. 1(a)), supporting the view that the abundance of CCG/CGG repeat was a specific feature of monocot genomes (Wang et al., Reference Wang, Li, Luo, Huang, Chen, Fang, Li, Chen and Zhang2011). For the di-nucleotide repeats, AG/CT motif was predominant and represented 32.39% of all of the characterized di-nucleotides (Fig. 1(b)).

Fig. 1. Frequency distribution of the tri- (a) and di-nucleotide (b) repeats identified in the transcriptome dataset.
A total of 584 primer pairs of genic SSR loci were successfully designed by ePrimer3 based on the obtained SSR-containing unigene sequences (online Supplementary Table S2). Subsequently, we randomly selected 36 primer pairs for the verification in eight individuals. Results indicated that 33 primer pairs (91.67%) successfully amplified DNA fragments (online Supplementary Fig. S2), with 21 (63.64%) of these producing a single product and the remaining 12 (33.33%; online Supplementary Table S3) primer pairs revealed allelic polymorphism. In comparison with previous studies (Liang et al., Reference Liang, Chen, Hong, Liu, Zhou, Li and Guo2009; Zheng et al., Reference Zheng, Pan, Diao, You, Yang and Hu2013) , our result yielded a similar rate of 60–90% amplification and the polymorphic ratio in the selected SSR primers was at a medium level when compared with some crops (Varshney et al., Reference Varshney, Sigmund, Börner, Korzun, Stein, Sorrells, Langridge and Graner2005b). The failure of amplification with some primer pairs could be due to the lack of primer specificity, presence of introns in the amplified sequence, variation in the number of repeated motifs, or assembly errors (Varshney et al., Reference Varshney, Graner and Sorrells2005a).
Additionally, to investigate whether these genic SSR markers developed for A. aequalis are available, the 12 polymorphic SSR markers were used to genotype a sample of 160 individuals from 20 natural populations of A. aequalis. Twelve primer pairs were all polymorphic; Na ranged from 2 to 5 alleles with a mean of 4.08, while PIC values ranged from 0.345 to 0.750 with an average value of 0.604 (Table 1). According to the previous report (Liu et al., Reference Liu, Xing, Zhang and Guo2015), the loci polymorphic level can be considered high, medium, or low if PIC > 0.5, 0.5 > PIC > 0.25 and PIC < 0.25. In this study, all loci in the 20 natural populations had a high PIC value (Mean = 0.604), indicating the utility of using these SSR markers for future genetic research. These results also indicated that the natural populations of A. aequalis maintain a high level of genetic diversity.
Table 1. Characteristics of the 12 polymorphic SSR markers

Na, number of alleles; Ne, effective number of alleles, Ho, observed heterozygosity, He, expected heterozygotes; I, Shannon's information index; PIC, polymorphic information content.
In summary, a comprehensive set of genic SSR markers was developed to greatly enrich the number of molecular markers available in A. aequalis, which will provide useful new resources for subsequent genetic studies involving comparative genomics, population structure, behavioural ecology and evolution of this highly noxious invasive weed.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S1479262118000138
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 31772181) and the Special Fund for Agro-scientific Research in the Public Interest (No. 201303031). We thank the editors and anonymous reviewers for useful and insightful comments on draft versions of the article.