INTRODUCTION
Human African trypanosomiasis (HAT), or ‘sleeping sickness’, results from infection with the protozoan parasites Trypanosoma brucei rhodesiense or T.b.gambiense. Both forms of HAT are fatal if untreated. There are two clinical stages associated with the disease. The first is the early or haemolymphatic stage where the parasites proliferate and spread in the blood and lymphatic system. This is followed by the late or CNS-stage of the disease which is marked by trypanosome invasion of the CNS. This pattern is found in both T.b.rhodesiense and T.b.gambiense infections; however T.b.rhodesiense is a more acute disease with rapid progression to the late stage whereas T.b.gambiense follows a more protracted course that can last several years before death occurs (Apted, Reference Apted and Mulligan1970). Once the parasites have established within the CNS many of the neurological features characteristic of the infection develop. Clinical manifestations suggestive of neurological involvement include mental and psychiatric disturbances and a wide range of motor symptoms, and signs of sensory involvement can become apparent. Visual impairment can also occur. In the latter stages of the disease the characteristic disruption of the normal sleep-wake cycle, from which the disease derives its popular name, develops with the presence of nocturnal insomnia and daytime somnolence, progressing to coma and ultimate death (Atouguia and Kennedy, Reference Atouguia, Kennedy, Davies and Kennedy2000). Treatment of early-stage disease is relatively effective; melarsoprol is the only drug available that can be used to treat both T.b.rhodesiense and T.b.gambiense infections once the CNS has become involved (Barrett et al. Reference Barrett, Boykin, Brun and Tidwell2007). Unfortunately melarsoprol treatment is associated with severe adverse reactions and can result in the generation of a post-treatment reactive encephalopathy (PTRE) in up to 10% of treated patients with a 50% mortality rate (Pepin and Milord, Reference Pepin and Milord1994). The mechanisms culminating in this severe adverse reaction remain unclear (Hunter and Kennedy, Reference Hunter and Kennedy1992; Kennedy, Reference Kennedy2006).
NEUROPATHOGENESIS
Pathological changes
The vast majority of the data on neuropathological changes associated with trypanosome infection and drug treatment, is derived from animal models of the disease with only a few post-mortem reports examining CNS material from human cases available. Many animal species have been used in investigations but most of the information comes from mouse, rat and primate models. In T.b.gambiense infections of vervet monkeys, the presence of perivascular cuffing, meningitis and encephalitis accompanied by an intense astocytosis has been reported. The inflammatory infiltrate was comprised of mononuclear cells, lymphocytes, plasma cells and immunoglobin containing Mott or morular cells (Ouwe-Missi-Oukem-Boyer et al. Reference Ouwe-Missi-Oukem-Boyer, Mezui-Me-Ndong, Boda, Lamine, Labrousse, Bisser and Bouteille2006). In experimental T.b.rhodesiense infections of vervet monkeys, a continuous chronic inflammatory process characterized by three distinct phases has been described. In phase 1, the animals developed a chronic meningitis with plasma cells, lymphocytes and monocytes in the subarachnoid and pial connective tissue. In phase 2, the neuroinflammation progressed from the meninges to the cerebral vessels, particularly those entering the brain, followed by the development of encephalitis in phase 3. In this model, trypanosomes appeared to spread together with the inflammatory cells, being first located in the choroid plexus, then spreading to the perivascular space and final to the brain parenchyma (Schmidt, Reference Schmidt1983). When mice were infected with T.b.rhodesiense a very similar pattern of neuroinflammation, characterized by the development of meningitis, perivascular cuffing and finally encephalitis, was described (Fink and Schmidt, Reference Fink and Schmidt1979). Again, the cell types involved were lymphocytes, plasma cells and macrophages with trypanosomes disseminating in concert with the inflammatory cell infiltrate. This inflammatory pattern was also echoed in a rat model of T.b.gambiense infection with the infiltration of plasma cells, Mott cells, lymphocytes and macrophages. In this study, a breakdown of T cell subsets showed that CD4+ cells outnumbered CD8+ cells in the infiltrate (Anthoons et al. Reference Anthoons, Van Marck, Gigase and Stevens1989). A very similar neuroinflammatory reaction developed following T.b.brucei infection of mice marked by the infiltration of lymphocytes, plasma cells, macrophages and Mott cells leading to a diffuse meningoencephalitis (Poltera et al. Reference Poltera, Hochmann, Rudin and Lambert1980). In the Glasgow model of HAT, mice infected with T.b.brucei GVR35 develop a mild meningitis as the infection enters the CNS-stage. This neuroinflammatory response can be exacerbated by sub-curative trypanocidal drug treatment to generate either a moderate neuroinflammatory reaction with the presence of inflammatory cells in the meninges and perivascular space or a severe meningoencephalitis involving infiltration of the neuropil by lymphocytes, plasma cells and macrophages (Fig. 1) (Hunter et al. Reference Hunter, Jennings, Kennedy and Murray1992b; Jennings et al. Reference Jennings, Gichuki, Kennedy, Rodgers, Hunter, Murray and Burke1997; Kennedy, Reference Kennedy1999). A marked astocytosis develops in parallel with the inflammatory cell infiltration and accompanying the onset of CNS disease (Fig. 2) (Hunter et al. Reference Hunter, Jennings, Kennedy and Murray1992b). An increased neuroinflammatory reaction as a result of sub-curative drug treatment is not an idiosyncrasy of our model since this phenomenon has been reported by several researchers (Schmidt and Sayer, Reference Schmidt and Sayer1982; Poltera et al. Reference Poltera, Sayer, Brighouse, Bovell and Rudin1985) and could relate to the development of the PTRE found in human cases of HAT (Hunter et al. Reference Hunter, Jennings, Adams, Murray and Kennedy1992a).
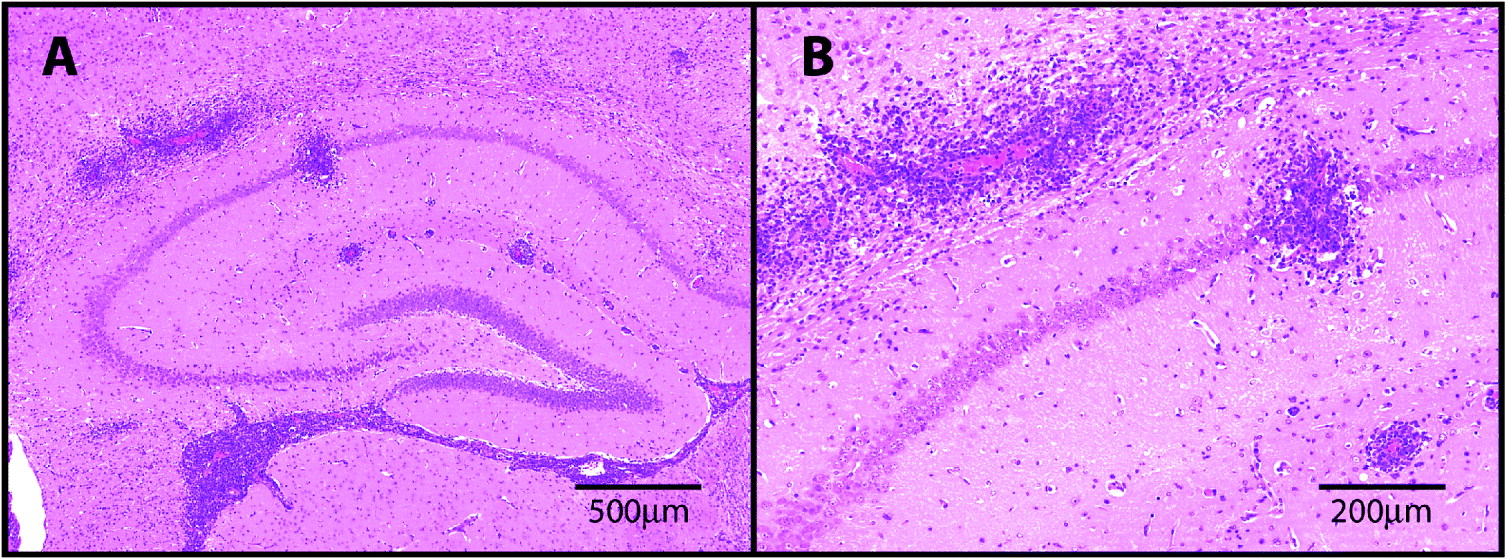
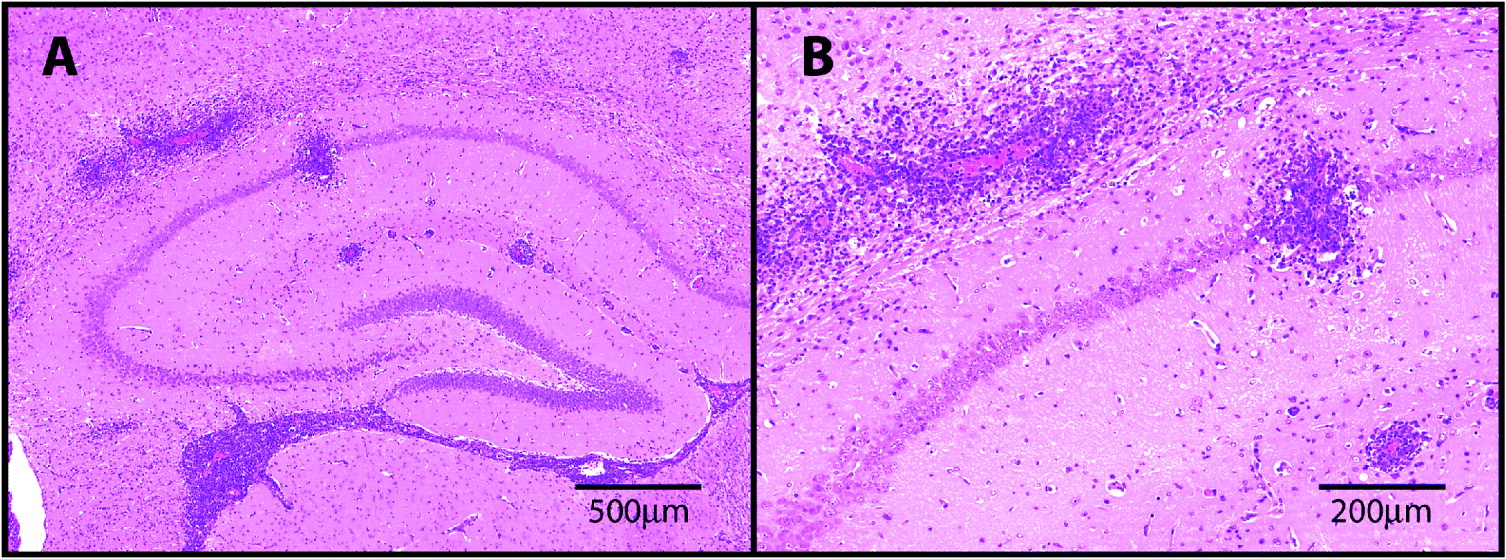
Fig. 1. H&E stained sections through the hippocampal brain region prepared from a mouse infected with T.b.brucei and treated sub-curatively with diminazene aceturate to exacerbate the neuroinflammatory response. Note the presence of severe perivascular cuffing and encephalitis (A). The extravasion of the inflammatory cells from the perivascular region into the parenchyma is shown in greater detail (B).

Fig. 2. Glial fibrillary acid protein (GFAP) staining of the hippocampal brain region prepared from a mouse infected with T.b.brucei and treated sub-curatively with diminazene aceturate to exacerbate the neuroinflammatory response. A wide spread astrocyte activation, present throughout the hippocampal region, is apparent (A). Note the large reactive astrocytes surrounding the inflamed blood-vessel and the prominent glia limitans (B).
In general, these models present a largely similar picture of the development of CNS disease with a stepwise infiltration of the brain by lymphocytes, plasma cells and macrophages (Kennedy, Reference Kennedy2006). Mott cells were frequently detected however neutrophils rarely appeared. Activation of microglia (Chianella et al. Reference Chianella, Semprevivo, Peng, Zaccheo, Bentivoglio and Grassi-Zucconi1999) and astrocytes (Hunter et al. Reference Hunter, Jennings, Kennedy and Murray1992b; Kennedy et al. Reference Kennedy, Rodgers, Jennings, Murray, Leeman and Burke1997; Ouwe-Missi-Oukem-Boyer et al. Reference Ouwe-Missi-Oukem-Boyer, Mezui-Me-Ndong, Boda, Lamine, Labrousse, Bisser and Bouteille2006) was frequently described. A general sparing of neuronal elements with little evidence of demyelination until the terminal stages of the infection are also a common feature (Fink and Schmidt, Reference Fink and Schmidt1979; Hunter et al. Reference Hunter, Gow, Kennedy, Jennings and Murray1991). Examination of human post-mortem material showed a similar pattern of neuroinflammation to that described in the animal models. Macrophages, lymphocytes and plasma cells were detected in the meninges, perivascular cuffs and infiltrating the neuropil. Mott cells were also present (Adams et al. Reference Adams, Haller, Boa, Doua, Dago and Konian1986; Adams and Graham, Reference Adams, Graham, Adams and Graham1998). Evidence of an inflammatory reaction was found only occasionally in the ventricles and choroid plexus (Adams et al. Reference Adams, Haller, Boa, Doua, Dago and Konian1986). Neuroinflammatory changes were noted in areas adjacent to the ventricles including the thalamus, hypothalamus and supraoptic nuclei as well as in the cerebral white matter (Adams et al. Reference Adams, Haller, Boa, Doua, Dago and Konian1986; Atouguia and Kennedy, Reference Atouguia, Kennedy, Davies and Kennedy2000). Diffuse microglial hyperplasia and astrocyte activation were also reported (Adams et al. Reference Adams, Haller, Boa, Doua, Dago and Konian1986; Pentreath, Reference Pentreath1989). In spite of the severity of the inflammatory reaction only nominal demyelination occurred with a general sparing of the nervous tissue (Bentivoglio et al. Reference Bentivoglio, Grassi-Zucconi, Olsson and Kristensson1994; Atouguia and Kennedy, Reference Atouguia, Kennedy, Davies and Kennedy2000). The PTRE is characterized by a marked increase in the severity of the reaction and in some instances can take the form of an acute haemorrhagic leucoencephalopathy (Adams et al. Reference Adams, Haller, Boa, Doua, Dago and Konian1986). Overall, the pathological changes reported following examination of human post-mortem samples are in accord with the data extrapolated from the animal models of CNS HAT and highlight the value of such model systems in pathological studies of this disease.
The role of CNS cytokines in the disease
Information on the expression of cytokines within the CNS during the course of the disease has been obtained through the use of animal models. Although these studies suggest an association between cytokine expression and the development of the neuroinflammation, care must be taken when interpreting the data to determine cause and effect in this highly complex system. Using comparative RT-PCR, up-regulated expression of TNF-α, MIP-1, IL-1α, IL-4, IL-6 and IFN-γ was found in the brains of T.b.brucei GVR35 infected mice (Hunter et al. Reference Hunter, Gow, Kennedy, Jennings and Murray1991). The induction of IL-6, MIP-1α, TNF-α and IFN-γ and up-regulation of IL-1α mRNA expression corresponded with the onset of astrocyte activation in this model whereas GM-CSF transcripts were only detected during the PTRE (Hunter et al. Reference Hunter, Jennings, Kennedy and Murray1992b). Protein levels of cytokines within the brain have been examined in a more recent study again utilising the T.b.brucei GVR35 mouse model of the disease. In these experiments, CNS levels of IFN-γ and TNF-α increased with the severity of the neuroinflammatory response while there was strong evidence of an exponential decline in the severity of the CNS reaction with increased levels of IL-10 and IL-6. A significant increase in IL-1β concentration in the brain was detected early after infection and remained high through the early and late-CNS stages of the disease although there was no correlation between IL-1β levels and the development of the neuroinflammatory response (Sternberg et al. Reference Sternberg, Rodgers, Bradley, MacLean, Murray and Kennedy2005). Intraventricular injection of IL-1 receptor antagonist in a rat model of trypanosome infection resulted in a restoration of body weight in the animals but had no effect on the neuropathology whereas injection of soluble TNF-α receptors ameliorated the CNS reaction (Quan et al. Reference Quan, He and Lai2003). These findings provide further evidence to support a correlation between TNF-α but not IL-1β concentrations in the severity of the neuroinflammatory reaction. In situ hybridisation (ISH) has also been employed to demonstrate the presence of a variety of inflammatory mediators in the brain of T.b.brucei-infected rats during the course of the infection (Quan et al. Reference Quan, Mhlanga, Whiteside, McCoy, Kristensson and Herkenham1999). IL-1β, TNF-α, IL-1β converting enzyme and inhibitory factor κBα (IκBα) mRNA was first detected in the median eminence, choroid plexus and arcuate nucleus eventually spreading throughout the parenchyma as the disease progressed while mRNA for IL-6 and IFN-γ was found most commonly in the choroid plexus. TUNEL staining showed the presence of apoptotic cells in the regions of cytokine over-expression suggesting a potentially cytotoxic action (Quan et al. Reference Quan, Mhlanga, Whiteside, McCoy, Kristensson and Herkenham1999). An increase in the number of cells expressing IFN-γ, TNF-α, TGF-β, and, to a lesser degree, IL-4 and IL-10, has also been detected in the rat brain by 6 hours following infection with a monomorphic strain of T.b.brucei (Sharafeldin et al. Reference Sharafeldin, Hamadien, Diab, Li, Shi and Bakhiet1999). This was paralleled by a significant rise in the level of IFN-γ and TGF-β found in the CSF. In a further study Sharafeldin et al. used immunocytochemistry to demonstrate increased staining of the chemokines; MIP-2, RANTES, MIP-1α and, to a lesser extent, MCP-1 in the brain early after infection (Sharafeldin et al. Reference Sharafeldin, Eltayeb, Pashenkov and Bakhiet2000). Initially astrocytes and microglia were the main source of these chemokines with T cells and macrophages taking over production later in the infection. This early production of chemokines by the resident glial cells suggests that the initial steps in the development of the CNS inflammatory disease could be controlled from within the CNS and that production of these factors may be responsible for initiating inflammatory cell infiltration. In both of these studies, the production of the inflammatory mediators was detected only hours following infection. It is possible that these changes are related to the extremely acute disease pattern associated with infections using this strain of trypanosome and therefore may not constitute a true reflection of the pathogenesis found in human disease.
In HAT patients, CSF concentrations of cytokines have been used to estimate the levels of these inflammatory mediators within the brain. Significant increases in the concentration of a number of cytokines and chemokines in the CSF have been detected in both T.b.rhodesiense and T.b.gambiense infections. In the Democratic Republic of Congo, significantly elevated levels of IL-6, IL-8 and IL-10 were found in the CSF of late-stage T.b.gambiense patients with the concentrations reducing after melarsoprol chemotherapy (Lejon et al. Reference Lejon, Lardon, Kenis, Pinoges, Legros, Bisser, N'Siesi, Bosmans and Buscher2002). Significant increases in CSF IL-10 and IL-6 levels have also been shown in T.b.rhodesiense infection in Uganda. Again, the concentration of these cytokines was reduced after drug treatment (MacLean et al. Reference MacLean, Odiit and Sternberg2001, Reference MacLean, Odiit and Sternberg2006). Further investigation of serum/CSF concentration quotients, indicated substantial intrathecal synthesis of IL-10 in 29% of patients, although the source of IL-6 could not be determined due to insufficient sample volume (MacLean et al. Reference MacLean, Odiit and Sternberg2006). In contrast to the cytokine profiles attained from rodent models of HAT, neither of these groups found significant changes in CSF TNF-α or IFN-γ concentrations (MacLean et al. Reference MacLean, Odiit and Sternberg2001, Reference MacLean, Odiit and Sternberg2006; Lejon et al. Reference Lejon, Lardon, Kenis, Pinoges, Legros, Bisser, N'Siesi, Bosmans and Buscher2002). However, evidence linking high plasma IFN-γ concentrations with disease progression to the CNS has been shown in a study examining the virulence of trypanosome infections in two distinct districts, Tororo and Soroti, in Uganda (MacLean et al. Reference MacLean, Odiit, MacLeod, Morrison, Sweeney, Cooper, Kennedy and Sternberg2007). It is possible that the apparent disparity between the CNS cytokine profiles found in the human disease and the mouse model reflect either differences between the cytokines present in the brain and the CSF or variations in the sensitivity of the assay systems used. Changes in chemokine concentrations have been found in studies conducted in Gabon and Angola (Courtioux et al. Reference Courtioux, Boda, Vatunga, Pervieux, Josenando, M'Eyi, Bouteille, Jauberteau-Marchan and Bisser2006). Here, CSF from late-stage patients infected with T.b.gambiense, was shown to contain elevated levels of MIP-1α, MCP-1, IL-8 and IL-1β. Furthermore, high concentration of these inflammatory mediators correlated with the presence of neurological signs including sleep, gait and sensory disturbance, abnormal reflexes and psychiatric disturbances.
Potential treatment adjuncts in HAT chemotherapy
There is an overwhelming need for immediate improvements in the chemotherapy of HAT. Therefore the use of drugs that are already licensed for other diseases may provide the most timely and cost-effective method to increase the safety and efficacy of existing trypanocidal therapies. Recent studies have shown that several compounds may be suitable for this purpose. Accumulating evidence suggests that the neuropeptide Substance P (SP) may play a role in the pathogenesis of a range of neurological disorders including late-stage trypanosomiasis (Kennedy et al. Reference Kennedy, Rodgers, Jennings, Murray, Leeman and Burke1997; Kennedy et al. Reference Kennedy, Rodgers, Bradley, Hunt, Gettinby, Leeman, de Felipe and Murray2003). Using a mouse model of late-stage HAT Kennedy et al. showed that administration of a highly selective non-peptide SP NK-1 receptor antagonist (RP,67-580, Rhone-Poulenc Rorer) precipitated a significant amelioration of the CNS inflammatory reaction and reduced the degree of astrocyte activation in treated animals compared to non-treated or enantiomer-treated controls. This indicates that SP may play a role in the generation of the neuroinflammatory reaction. A humanised SP receptor antagonist, aprepitant (EMEND® Merck & Co. Inc.) has been tested in the HAT mouse model in conjunction with melarsoprol treatment (Rodgers et al. Reference Rodgers, Bradley and Kennedy2007). No additional or unexpected CNS reactions were apparent indicating that the use of aprepitant as an adjunct to melarsoprol administration may be possible.
The second generation tetracycline antibiotic, minocycline, has also shown potential in this area. Minocycline treatment impedes the passage of leucocytes into the CNS when administered in experimental autoimmune encephalitis indicating that it may have therapeutic potential in multiple sclerosis. When minocycline was given to mice infected with T.b.brucei, beginning on the day of parasite inoculation, both leucocyte and trypanosome transmigration into the brain parenchyma were reduced together with astrocyte and microglial activation (Masocha et al. Reference Masocha, Rottenberg and Kristensson2006). This was accompanied by a decrease in the expression of adhesion molecules; ICAM-1 and E-selectin, matrix metalloproteases; MMP-3, MMP-8 and MMP-12 and cytokines; TNF-α, IFN-γ, IL-1α, IL-1β and IL-6 as measured by comparative real-time reverse transcription PCR (Masocha et al. Reference Masocha, Rottenberg and Kristensson2006). Furthermore, when minocycline was administered in combination with the trypanocidal drug suramin, given at a sub-curative dose, an extended aparasitaemic period was achieved (Amin et al. Reference Amin, Masocha, Ngan'dwe, Rottenberg and Kristensson2008). Although more work is required in this area it may be possible to use minocycline as an adjunct to early-stage drugs to prevent relapse in ‘intermediate’ stage disease. The presence of this ‘intermediate’ or ‘early late-stage’ of infection was suggested following successful treatment of patients presenting with trypanosomes in their CSF and white blood cell counts of no higher than 20, with the early stage drug pentamidine (Doua et al. Reference Doua, Miezan, Sanon, Boa and Baltz1996; Lejon et al. Reference Lejon, Legros, Savignoni, Etchegorry, Mbulamberi and Buscher2003a, Reference Lejon, Reiber, Legros, Dje, Magnus, Wouters, Sindic and Buscherb). These criteria are above the threshold figure of 5 white blood cells suggest by the WHO guidelines to define CNS-stage disease (WHO, 1998).
The kynurenine pathway, the major pathway in the metabolism of tryptophan, has been little explored with regard to trypanosome infections. There are numerous catabolites formed along this pathway, some have neuroprotective properties while others are neurotoxic. A recent study employing an experimental compound, Ro-61-8048, that inhibits kynurenine-3-monooxygenase and manipulates the pathway towards the production of neuroprotective kynurenic acid found a reduction in the severity of the neuroinflammatory response in mice infected with T.b.brucei following inhibitor treatment (Rodgers et al. Reference Rodgers, Stone, Barrett, Bradley and Kennedy2009). These findings suggested that the kynurenine pathway is a valid target for future investigations in the development of novel chemotherapeutic adjuncts.
Magnetic Resonance Imaging (MRI)
Although sophisticated neuroimaging techniques are generally unavailable in HAT-endemic areas, on a few occasions the increase in international travel has provided an opportunity to use MRI to investigate the CNS-stage of the disease. Using this technology, symmetrical focal lesions in the deep white matter of the internal capsules, cerebellum and splenium of the corpus callosum (Sabbah et al. Reference Sabbah, Brosset, Imbert, Bonardel, Jeandel and Briant1997) as well as abnormalities in the basal ganglia, external capsule and extreme capsule (Gill et al. Reference Gill, Chatha and Carpio-O'Donovan2003) were detected. The possible use of MRI to distinguish between relapsed CNS disease and the development of the PTRE has also been reported following scanning of a comatose HAT patient receiving melarsoprol treatment (Braakman et al. Reference Braakman, van de Molengraft, Hubert and Boerman2006). In this case, MRI showed the presence of multiple white matter lesions and lesions in the central grey matter and cortex. Fluid-attenuated inversion recovery T2-weighted MRI (T2*) did not reveal haemosiderin within these areas indicating that the lesions were not attributable to the presence of microhaemorrhages (Braakman et al. Reference Braakman, van de Molengraft, Hubert and Boerman2006). In another case, a patient became confused and apraxic followed by the development of generalised tonic-clonic seizures during a course of melarsoprol (Checkley et al. Reference Checkley, Pepin, Gibson, Taylor, Jager and Mabey2007). MRI investigation showed widespread bilateral abnormalities involving the supratentorial and infratentorial white matter in T2-derived images. In some of these areas, including the midbrain and brainstem, multiple microhaemorrhages were detected following T2* examination suggestive of the PTRE (Checkley et al. Reference Checkley, Pepin, Gibson, Taylor, Jager and Mabey2007). Although the data presented within these studies indicate that MRI can provide valuable information in the diagnosis and management of the PTRE, it is highly unlikely that this technology, while commonplace in the developed world, will become available to the majority of those patients affected by HAT.
TRYPANOSOMES IN THE BRAIN
Although CNS involvement in HAT is well recognised in the progress of the disease there have been few reports of parasites detected within the brain (Mott, Reference Mott1907). This could be the result of inadequate fixation of the brain allowing degradation of the parasites before examination of the tissue or trypanocidal drug treatment prior to the death of the patient clearing the parasite from the brain (Calwell, Reference Calwell1937). Many questions therefore still exist regarding the precise route and mechanisms used by the trypanosomes to facilitate CNS invasion.
Blood-brain barrier systems
The concept of the brain as an ‘immuno-privileged site’, with little or no capacity to mount an immune response, has altered drastically in recent times. Effective immune surveillance within the CNS, producing a parenchymal immune-mediated reaction, has been reliably demonstrated following peripheral immunisation with autoantigens or pathogens sequestered within the brain and the passage of leucocytes into the CNS is now an accepted process (Man et al. Reference Man, Ubogu and Ransohoff2007). However, in order to preserve normal brain functions it is essential to maintain the specialized nature of the environment present within the CNS. Therefore barrier mechanisms exist to control the trafficking of cells and most molecules between the peripheral and central compartments. These barriers prevent or severely impede this exchange and exist in three forms; the blood-brain barrier (BBB), the blood-CSF barrier and the arachnoid barrier. These have been described in detail elsewhere and only a brief description will be given in this article (Abbott et al. Reference Abbott, Ronnback and Hansson2006; Man et al. Reference Man, Ubogu and Ransohoff2007; Saunders et al. Reference Saunders, Ek, Habgood and Dziegielewska2008). The BBB is situated between the lumen of the cerebral blood vessels and the brain parenchyma. The physical barrier is formed by the endothelial cells comprising the vessels walls. These endothelial cells are held in close apposition to each other via tight junctions formed by four trans-membrane proteins, occludin, claudins, junctional adhesion proteins (JAMs) and endothelial selective adhesion molecules (ESAMs) together with a number of cytoplasmic accessory molecules (Abbott et al. Reference Abbott, Ronnback and Hansson2006). Adherens junctions are also present between the endothelial cells and stabilize the cell-cell interactions in the junctional zone (Abbott et al. Reference Abbott, Ronnback and Hansson2006). The endothelial cells are surrounded by basement membrane and astrocyte foot processes, which form the glia limitans. Pericytes lie within the basement membrane between the endothelial cells and the astrocytes. These cells have long cytoplasmic processes that encircle the endothelial cells and make contact with them through specialized junctions (Zlokovic, Reference Zlokovic2008). In addition, neurons and microglia found adjacent to the astrocytes are thought to be important in barrier maintenance. These components collectively form the neurovascular units that constitute the classic BBB (Hawkins and Davis, Reference Hawkins and Davis2005; Abbott et al. Reference Abbott, Ronnback and Hansson2006). This barrier prevents paracellular crossing of most molecules with the exception of oxygen and carbon dioxide. Small lipophilic molecules, such as barbiturates, or ethanol can also diffuse across the barrier; movement of most other molecules requires specific transport systems present on the luminal and abluminal surfaces of the cells. There are a variety of pathways available to allow the exchange of molecules including specific transport proteins, receptor-mediated endocytosis and adsorptive mediated endocytosis (Abbott et al. Reference Abbott, Ronnback and Hansson2006).
The blood-CSF barrier is found between the choroid plexus blood vessels and the CSF. The choroid plexus is a highly vascularised tissue located in the cerebral ventricles. In contrast to the brain parenchyma, the blood vessels here are fenestrated and the tight junctions linking the endothelial cells are discontinuous; therefore these cells form a non-restrictive barrier. Tight junctions do exist between the epithelial cells of the choroid plexus and these cells effectively constitute the blood-CSF barrier (Johansson et al. Reference Johansson, Dziegielewska, Liddelow and Saunders2008).
The arachnoid barrier is probably the least familiar of the three barrier types. This barrier is situated between the dura and the pia and appears to be structurally the most complex of the three. The blood vessels entering the dura are fenestrated and as such form a poor barrier. However, the endothelial cells of the blood vessels entering the arachnoid and the pia are connected by tight junctions in a similar fashion to those found in the parenchyma although they lack the presence of astrocytes and pericytes. In addition, there are tight junctions between the epithelial cells forming the basal layer of the arachnoid membrane and this layer acts as a barrier between the dura and the CSF filled sub-arachnoid space. It is worthy of note that neurons are typically no more than 8–20 μm from a brain capillary but this distance can increase to centimetres when considering neuronal proximity to CSF compartments. This fact highlights the importance of the classical BBB, or neurovascular unit, in comparison with the blood-CSF and arachnoid barriers in the maintenance of the CNS microenvironment (Abbott et al. Reference Abbott, Ronnback and Hansson2006; Zlokovic, Reference Zlokovic2008).
Leucocyte transmigration into the brain can occur through these barriers and involves a complex series of interactions between the leucocytes and the epithelial cells. The process is initiated when the leucocytes form loose connections with the endothelial cells via selectin-integrin interactions. This loose binding or ‘tethering’ allows the leucocyte to ‘roll’ along the endothelial cell barrier with the flowing blood testing for the presence of surface-bound luminal chemokines. When these chemokines are encountered by the leucocyte chemokine receptors signalling pathways within the leucocyte are activated precipitating conformational changes in the leucocyte integrins leading to high-affinity binding to the endothelial cell typically via adhesion molecules. The leucocytes then move to the inter-endothelial junction where they extend protrusions through the junction sampling for abluminal chemokines. Transmigration of the leucocyte occurs in response to the presence of these chemokines following a chemotactic gradient. Once the cells have crossed the endothelial cell layer they are sequestered in the perivascular space between the endothelial cell basement membrane and the parenchymal basement membrane. Completion of the transmigration into the brain parenchyma requires the action of matrix metalloproteinases (MMP's) that degrade the cellular matrix and facilitate leucocyte passage through the basement membrane (Man et al. Reference Man, Ubogu and Ransohoff2007).
Trypanosome distribution in the brain
Parasites have been detected in the brain early after infection with T.b.brucei in rodent models (Schultzberg et al. Reference Schultzberg, Ambatsis, Samuelsson, Kristensson and Van Meirvenne1988; Masocha et al. Reference Masocha, Robertson, Rottenberg, Mhlanga, Sorokin and Kristensson2004). In a rat model of T.b.brucei infection, trypanosomes were first detected in the spinal and trigeminal ganglia, the median eminence and hypothalamic area as well as the neural lobe of the pituitary. The stroma of the choroid plexus was also highly parasitized. As the infection progressed, the parasites could be detected in the pineal gland and the area postrema. Trypanosomes were only rarely detected in the parenchyma until the terminal stage of the infection when many parasites were found in the brain parenchyma and in the spinal cord (Schultzberg et al. Reference Schultzberg, Ambatsis, Samuelsson, Kristensson and Van Meirvenne1988). It has been suggested that trypanosome infection causes progressive damage to the BBB as the disease advances (Pentreath et al. Reference Pentreath, Baugh and Lavin1994; Philip et al. Reference Philip, Dascombe, Fraser and Pentreath1994). To investigate this, fluorescent dye was injected into the jugular vein of T.b.brucei infected rats. The dye was first detected in the thalamus and hypothalamus. At later time points the dye was found permeating the white and grey matter of the cortex indicating a progressive loss in BBB integrity as the disease developed (Philip et al. Reference Philip, Dascombe, Fraser and Pentreath1994). Trypanosomes were also detected in the brain parenchyma. A later study, again utilising T.b.brucei infection of rats, showed large numbers of parasites in the choroid plexus, median eminence and pineal gland (Mulenga et al. Reference Mulenga, Mhlanga, Kristensson and Robertson2001). Trypanosomes were also found in the meninges but only rarely in the sub-pial tissue. The numbers of parasite increased as the disease progressed and their distribution spread to the brain parenchyma. No disruption to the tight junctions of the BBB was found following staining with occludin and ZO-1 indicating that the trypanosomes had entered the brain without triggering a permanent breakdown in the integrity of the BBB tight junctions (Mulenga et al. Reference Mulenga, Mhlanga, Kristensson and Robertson2001). This suggests that the trypanosomes enter the CNS in a regulated fashion and do not simply diffuse into the CNS as a consequence of barrier breakdown. Furthermore, studies by Masocha and colleagues found early invasion of the circumventricular organs in rats and mice following infection with T.b.brucei. From here the trypanosomes spread to the parenchyma via intracerebral vessels rather than through CSF (Masocha et al. Reference Masocha, Robertson, Rottenberg, Mhlanga, Sorokin and Kristensson2004). Areas such as the circumventricular organs contain neurons that are specialized in neurosecretion or chemosensitivity; therefore the endothelium in these areas is leaky to allow tissue blood exchange of molecules. These areas are separated from the rest of the brain by a glial barrier and a barrier at the ependyma isolates them from the CSF (Abbott et al. Reference Abbott, Ronnback and Hansson2006). This could explain the apparent delay in parenchymal invasion until the later stages of the infection and equate to the recently suggested ‘early-late-stage’ of the disease in human infection (Doua et al. Reference Doua, Miezan, Sanon, Boa and Baltz1996; Lejon et al. Reference Lejon, Legros, Savignoni, Etchegorry, Mbulamberi and Buscher2003a, Reference Lejon, Reiber, Legros, Dje, Magnus, Wouters, Sindic and Buscherb; Kennedy, Reference Kennedy2004).
There is also evidence to suggest that the pro-inflammatory cytokine IFN-γ may play a significant role in trypanosome invasion of the CNS. In experiments utilising IFN-γ or IFN-γ receptor-deficient mice, a reduced number of trypanosomes was detected within the CNS compared to their wild-type counter-parts (Masocha et al. Reference Masocha, Robertson, Rottenberg, Mhlanga, Sorokin and Kristensson2004). In these knockout mice, the parasites traversed the epithelial cell layer but failed to penetrate the parenchymal basement membrane. Whether administration of IFN-γ to the IFN-γ knockout strain would allow the trypanosomes to cross into the brain remains to be determined. Studies also indicate that the laminin composition of the basement membrane plays an important role in trypanosome transmigration into the CNS (Masocha et al. Reference Masocha, Robertson, Rottenberg, Mhlanga, Sorokin and Kristensson2004, Reference Masocha, Rottenberg and Kristensson2007). Masocha et al. have shown that basement membrane containing laminin α4 is permissive to trypanosome transmigration into the brain while that containing laminin α5 is inhibitory to parasite entry of the parenchyma (Masocha et al. Reference Masocha, Robertson, Rottenberg, Mhlanga, Sorokin and Kristensson2004) mirroring the restrictions seen in T cell transmigration into the CNS (Sixt et al. Reference Sixt, Engelhardt, Pausch, Hallmann, Wendler and Sorokin2001). These studies also demonstrated that trypanosomes fail to reach the CNS in infections of recombinant activating gene (RAG)-1-deficient mice, which lack both T and B cells, strengthening the hypothesis that lymphocyte transmigration plays a crucial role in the ability of the trypanosomes to penetrate the brain parenchyma. Studies investigating the effects of minocycline in trypanosome infections, as described above, provide further evidence linking trypanosome and T cell transmigration into the CNS (Masocha et al. Reference Masocha, Rottenberg and Kristensson2006, Reference Masocha, Rottenberg and Kristensson2007; Amin et al. Reference Amin, Masocha, Ngan'dwe, Rottenberg and Kristensson2008).
In vitro BBB studies
It is now possible to mimic the BBB using in vitro BBB systems and these can be useful tools in identifying the cellular and molecular pathways important to trypanosome penetration of the brain. In these systems, brain microvascular endothelial cells (BMECs) prepared from various species, including humans, can be grown on collagen-coated membranes that remain suspended in culture medium. This effectively isolates the fluid in the upper chamber, equivalent to the lumen of the blood vessel, from that in the bottom of the well, mimicking the abluminal surface of the blood vessel. The transwell system allows agents to be added to either side of the barrier and their ability to penetrate to the opposite side assessed. In addition, the effect of the agent on the real time transendothelial electrical resistance (TEER) can be monitored to measure the effect of the agent on the tightness of the barrier (Grab and Kennedy, Reference Grab and Kennedy2008). Studies utilising this system to investigate the interaction of trypanosomes with the endothelial cells have suggested that T.b.rhodesiense [originally classified as T.b.gambiense (Nikolskaia et al. Reference Nikolskaia, de A. Lima, Kim, Lonsdale-Eccles, Fukuma, Scharfstein and Grab2008)] appears to cross the human BMECs more readily than T.b.brucei. Data from multiple experiments found that 18·7% of the total T.b.rhodesiense inoculate traversed the barrier compared to 2·8% of the T.b.brucei inoculate (Grab et al. Reference Grab, Nikolskaia, Kim, Lonsdale-Eccles, Ito, Hara, Fukuma, Nyarko, Kim, Stins, Delannoy, Rodgers and Kim2004). However, further investigation broadening the range of strains tested is required as these attributes could be peculiar to the individual strains included in this study. Only a transient reduction in TEER was seen between 3 and 6 hours following introduction of either T.b.rhodesiense or T.b.brucei to the transwell culture system suggesting that the parasites do not penetrate the cell layer through damaging the barrier integrity. Fluorescence microscopy demonstrated that the trypanosomes attach to the BMECs at the inter-endothelial cell borders (Grab et al. Reference Grab, Nikolskaia, Kim, Lonsdale-Eccles, Ito, Hara, Fukuma, Nyarko, Kim, Stins, Delannoy, Rodgers and Kim2004). Further studies, employing confocal microscopy, have shown the presence of trypanosomes within the endothelial cell. Neither the viability nor the significance of these intracellular forms is currently known (Nikolskaia et al. Reference Nikolskaia, Kim, Kovbasnjuk, Kim and Grab2006b). The importance of parasite-derived substances in the ability of trypanosomes to traverse BMECs has been demonstrated in a recent study investigating trypanosome cysteine protease enzymes (Nikolskaia et al. Reference Nikolskaia, de A. Lima, Kim, Lonsdale-Eccles, Fukuma, Scharfstein and Grab2006a). These enzymes belong to the papain family and are important for the growth and survival of a number of protozoan pathogens (Sajid and McKerrow, Reference Sajid and McKerrow2002). Two forms of cysteine protease, cathepsin B-like enzyme and cathepsin L-like enzyme, also known as brucipain, are present in T. brucei. Transient alterations in intracellular calcium concentrations ([Ca2+]i) were detected in human BMECs exposed to either trypanosomes or trypanosome- conditioned medium. If a specific cathespin L-like cysteine protease inhibitor K11777 was added to the culture medium prior to exposure to the parasites the changes in [Ca2+]i were prevented. This was not the case when cathepsin B-like cysteine protease inhibitors were introduced to the medium indicating a causal role for brucipain in the [Ca2+]i fluctuations. The importance of the [Ca2+]i changes in parasite traversal of the barrier was demonstrated in a series of experiments using BMECs pretreated with either a [Ca2+]i chelator (BAPTA-AM), a phospholipase C inhibitor (U73122) or a protein kinase C inhibitor (Calphostin C) to prevent alteration in the [Ca2+]i. Use of these compounds precluded trypanosome crossing of the BMECs in a dose-dependent fashion. Additionally, enhanced crossing of the barrier was found if brucipain-enriched culture medium was added to the system again indicating the central role of brucipain in trypanosome penetration of the in vitro BBB system (Nikolskaia et al. Reference Nikolskaia, de A. Lima, Kim, Lonsdale-Eccles, Fukuma, Scharfstein and Grab2006a). The potential functions of cathepsin-L and cathepsin-B were further investigated using trypanosomes electroporated with plasmids containing either brucipain or cathepsin-B transgenes designed to produce RNAi following induction with tetracycline. These parasites were used in murine infections. Induction of RNAi to inhibit cathepsin-L (brucipain) extended the survival period of 50% of the animals compared with non-induced controls and had no effect on the parasitaemia. However, induction of RNAi targeting of cathepsin-B cleared the parasites from the bloodstream and prevented a lethal infection with survival of the animals until the endpoint of the experiment. These findings indicate the cathepsin-B may be a promising chemotherapeutic target to attain curative treatments while cathepsin-L may be play a role in disease progression to the CNS (Abdulla et al. Reference Abdulla, O'Brien, Mackey, Sajid, Grab and McKerrow2008).
Sleep disturbances
The most commonly recognised clinical symptom associated with late-stage HAT is an alteration in sleep patterns, with the presence of daytime somnolence and night-time insomnia, which occurs following infection and becomes more pronounced as the infection advances (Buguet et al. Reference Buguet, Bisser, Josenando, Chapotot and Cespuglio2005). Indeed these symptoms led to the disease becoming commonly known as ‘sleeping sickness’.
HAT and circadian rhythms
In HAT patients, alterations in the normal circadian variation of growth hormone secretion, prolactin and cortisol levels and plasma renin activity have been described (Radomski et al. Reference Radomski, Buguet, Bogui, Doua, Lonsdorfer, Tapie and Dumas1994; Lundkvist et al. Reference Lundkvist, Kristensson and Bentivoglio2004). The master circadian pacemaker, responsible for the control of these endogenous recurring rhythms in mammals, is localised to the suprachiasmatic nuclei (SCN) situated in the anterior, ventral hypothalamus of the brain (Lundkvist et al. Reference Lundkvist, Kristensson and Bentivoglio2004; Coogan and Wyse, Reference Coogan and Wyse2008). The rhythms controlled by the SCN are entrained through light signals conducted to the SCN via the neurons of retino-hypothalamic tract. In normal rats, light stimulation produces a rapid expression of c-fos in the SCN but this induction is strikingly reduced in T.b.brucei infected rats (Peng et al. Reference Peng, Kristensson and Bentivoglio1994). Although the retino-hypothalamic tract remains intact in these animals, a decrease in the expression of glutamate receptor sub-units in the area of the SCN innervated by the retinal fibres has been demonstrated (Lundkvist et al. Reference Lundkvist, Christenson, ElTayeb, Peng, Grillner, Mhlanga, Bentivoglio and Kristensson1998a). In addition, a decrease in the frequency but not the amplitude of excitatory post-synaptic activity, an event normally under circadian control, has been detected in the SCN in brain slices prepared from T.b.brucei-infected rats (Lundkvist et al. Reference Lundkvist, Hill and Kristensson2002). Furthermore, Lundkvist et al. demonstrated that this reduction in firing frequency could be mimicked by treatment of brain slices prepared from normal rats with a cocktail containing the pro-inflammatory cytokines IFN-γ and TNF-α together with LPS (Lundkvist et al. Reference Lundkvist, Hill and Kristensson2002). The influence of the immune system in the control of the circadian clock has been comprehensively reviewed elsewhere (Coogan and Wyse, Reference Coogan and Wyse2008) and diurnal variation in the expression of cytokines including IL-1β, TNF-α, and IFN-γ as well as TNF-α receptors and IFN-γ receptors within the brain and SCN have been described together with the rhythmic expression of the cell signalling molecules JAK1, JAK2 and STAT1 (Lundkvist et al. Reference Lundkvist, Robertson, Mhlanga, Rottenberg and Kristensson1998b; Coogan and Wyse, Reference Coogan and Wyse2008). Many of these pro-inflammatory mediators are highly expressed in HAT and animal models of the disease, as described above. Moreover, IL-1β and TNF-α in particular are known to influence sleep (Krueger et al. Reference Krueger, Obal, Fang, Kubota and Taishi2001; Obal and Krueger, Reference Obal and Krueger2003; Opp, Reference Opp2005).
HAT and sleep structure
In addition to changes in circadian sleep-wake profiles, alterations in the sleep structure have been described in late-stage HAT patients (Montmayeur et al. Reference Montmayeur, Brosset, Imbert and Buguet1994; Buguet et al. Reference Buguet, Bourdon, Bouteille, Cespuglio, Vincendeau, Radomski and Dumas2001, Reference Buguet, Bisser, Josenando, Chapotot and Cespuglio2005) and animal models of the disease (Toth et al. Reference Toth, Tolley, Broady, Blakely and Krueger1994; Grassi-Zucconi et al. Reference Grassi-Zucconi, Semprevivo, Mocaer, Kristensson and Bentivoglio1996). Under normal conditions, sleep can be divided into two sequential phases; REM (rapid eye movement) sleep and non-REM or slow-wave sleep (SWS). REM sleep is connected with vivid dreaming while non-REM is associated with reduced neuronal activity. On falling asleep, individuals generally enter a non-REM stage followed by a period of REM sleep. This cycle repeats throughout the night (McCarley, Reference McCarley2007). In polysomnographs of late-stage HAT patients, striking changes to this pattern are seen with the frequent occurrence of sleep on-set REM (SOREM) where patients go from wakefulness straight into REM sleep without passing through a preceding non-REM stage (Buguet et al. Reference Buguet, Bourdon, Bouteille, Cespuglio, Vincendeau, Radomski and Dumas2001, Reference Buguet, Bisser, Josenando, Chapotot and Cespuglio2005). Polysomnography has also confirmed that the total time spent asleep, and the duration of each of the sleep stages, remain largely unaltered in HAT patients irrespective of disease severity (Buguet et al. Reference Buguet, Bourdon, Bouteille, Cespuglio, Vincendeau, Radomski and Dumas2001). Trypanosome infections therefore result in a dysregulation and fragmentation of sleep patterns rather than hypersomnia. Changes in EEG events are also evident in late-stage HAT such as the slowing of the EEG or the presence of periodic slow waves during periods of wakefulness. Altered K complexes, degraded spindals, hypersynchronic slow waves and hypnopomic delta bursts occurring during slow wave sleep have been described (Buguet et al. Reference Buguet, Bisser, Josenando, Chapotot and Cespuglio2005). These abnormalities ameliorate following trypanocidal chemotherapy. The potential use of polysomnography, both as a diagnostic tool to distinguish between early and late-stage disease and to monitor the efficacy of chemotherapy, has been suggested although further studies in this area are required (Buguet et al. Reference Buguet, Bisser, Josenando, Chapotot and Cespuglio2005). In a rat model of trypanosomiasis, the onset of sleep changes characterized by numerous awakenings from slow-wave sleep, a reduction in the average length of slow-wave sleep periods and reduced REM sleep latency were predictive of the terminal stage of the infection (Bentivoglio et al. Reference Bentivoglio, Grassi-Zucconi, Olsson and Kristensson1994; Grassi-Zucconi et al. Reference Grassi-Zucconi, Harris, Mohammed, Ambrosini, Kristensson and Bentivoglio1995). Significant decreases in slow-wave sleep and delta wave amplitude during slow-wave sleep were also apparent in a rabbit model of HAT (Toth et al. Reference Toth, Tolley, Broady, Blakely and Krueger1994).
The initial invasion of the brain by accumulations of trypanosomes is thought to occur in regions including the circumventricular organs and choroid plexus as described above. These areas lie in close proximity to the SCN and sites involved in sleep regulation (Bentivoglio and Kristensson, Reference Bentivoglio and Kristensson2007) raising the possibility that host parasite interactions could result in the production of mediators such as cytokines or neurotransmitters that can act on these brain regions to alter both the circadian control of sleep-wake cycles and sleep structure itself.
CONCLUDING REMARKS
The interactions between trypanosomes, the host immune system and the CNS are numerous and complex. In recent years, some progress has been made in unravelling the mysteries of the disease but many questions still remain. Further studies investigating the apparent link between inflammatory cell and trypanosome transmigration across the BBB into the CNS are required to elucidate molecules either suitable for drug targeting or to provide markers for disease progression. The application of advanced molecular analysis techniques to define these host-parasite interactions, with the ultimate goal of manipulating the response to a more favourable outcome, provides the basis for significant advances in HAT research in the coming years.
ACKNOWLEDGEMENTS
I am grateful to Professor Peter Kennedy for his enthusiasm in pursuing trypanosomiasis research, helpful comments and critical review of this manuscript. Our continued research investigating the pathogenic mechanisms involved in CNS-stage HAT is supported by funding from the Wellcome Trust [082786] and the MRC [G0601059].