INTRODUCTION
In the last 15 years, Drosophila melanogaster has become an important model of the genetics and evolution of innate immunity and host-parasite interactions (Hoffmann, Reference Hoffmann2003). However, much of this research relies on microbes that would never infect flies in natural populations, and we remain remarkably ignorant of the natural parasites and pathogens of Drosophila. For example, the immune response of D. melanogaster to trypanosomatids was studied using Crithidia bombi and C. fasciculata, which naturally infect bumblebees and mosquitoes respectively (Boulanger et al. Reference Boulanger, Ehret-Sabatier, Brun, Zachary, Bulet and Imler2001). The best-described pathogens of Drosophila are viruses, with numerous different families of viruses having been found infecting flies in the wild (Brun and Plus, Reference Brun, Plus, Ashburner and Wright1980; Longdon et al. Reference Longdon, Obbard and Jiggins2010). Recently, bacterial pathogens have also been isolated from wild flies (Corby-Harris et al. Reference Corby-Harris, Pontaroli, Shimkets, Bennetzen, Habel and Promislow2007; Juneja and Lazzaro, Reference Juneja and Lazzaro2009). However, there has been very little research into the protozoan parasites of Drosophila for the last 40 years (see Ebbert et al. (Reference Ebbert, Marlowe and Burkholder2003) for exceptions).
Trypanosomatids (Kinetoplastida: Trypanosomatidae) are flagellate protozoans that infect a wide range of invertebrates, vertebrates and plants. The dixenous (2-host) species are best known, as they include insect-vectored parasites of vertebrates. The monoxenous species, which are restricted to insects, are less well studied. The large majority of trypanosomatids that have been reported have been found in the Diptera (flies) and Hemiptera (bugs) (Podlipaev, Reference Podlipaev2001), and in these groups they can be very common. For example, a recent survey of 170 species of insects from the suborder Heteroptera found that 9% of species carried trypanosomatids, with an overall prevalence of 22% (Maslov et al. Reference Maslov, Westenberger, Xu, Campbell and Sturm2007). However, despite these recent studies the true diversity of trypanosomatids is unknown (Podlipaev, Reference Podlipaev2001). The first records of trypanosomatids in Drosophila were made over 100 years ago in Drosophila confusa (Chatton and Alilaire, Reference Chatton and Alilaire1908), and they have subsequently been discovered in a range of other species (Ebbert et al. Reference Ebbert, Burkholder and Marlowe2001). In a study of 8 species of North American Drosophila the prevalence of infection ranged from 1to17% (Ebbert et al. Reference Ebbert, Burkholder and Marlowe2001), although there have been reports of the prevalence reaching 40% in D. melanogaster (McGhee and Cosgrove, Reference McGhee and Cosgrove1980). The trypanosomatids infect the gut, and transmission occurs when food is contaminated with faeces or with the cadavers of infected flies (Rowton and McGhee, Reference Rowton and McGhee1983). Although trypanosomatids can be transmitted between distantly related species of Drosophila in the laboratory, rates of transmission in a new host can be much reduced suggesting that there is some degree of host specificity (McGhee et al. Reference McGhee, Hanson and Schmittner1969). Trypanosomatid infections are harmful to the host, as infected D. melanogaster have significantly longer development times than uninfected individuals (Ebbert et al. Reference Ebbert, Marlowe and Burkholder2003).
Until recently the only way to identify trypanosomatids and study their diversity was to either conduct detailed studies in culture or use microscopy to describe their morphology, but both of these approaches have their limitations. Culture-based methods tend to miss much of the diversity because many trypanosomatids cannot be cultivated (Yurchenko et al. Reference Yurchenko, Lukes, Jirku and Maslov2009). Additionally, insect trypanosomatids are difficult to distinguish morphologically and phylogenies based on morphological characters have proved unreliable (Podlipaev et al. Reference Podlipaev, Sturm, Fiala, Fernanades, Westenberger, Dollet, Campbell and Lukes2004). PCR and DNA sequencing can avoid these problems, and these approaches have now shown that insect trypanosomatids are highly diverse and have provided detailed phylogenies (Podlipaev et al. Reference Podlipaev, Sturm, Fiala, Fernanades, Westenberger, Dollet, Campbell and Lukes2004; Yurchenko et al. Reference Yurchenko, Lukes, Xu and Maslov2006; Maslov et al. Reference Maslov, Westenberger, Xu, Campbell and Sturm2007; Votypka et al. Reference Votypka, Maslov, Yurchenko, Jirku, Kment, Lun and Lukes2010). The aim of this study was to use these PCR-based approaches to study trypanosomatids that infect Drosophila. To assess the diversity of Drosophila trypanosomatids, we sequenced the spliced leader RNA gene. Highly conserved exon sequences allow conserved primers to be designed that will amplify this gene from all the genera of trypanosomatids, while a hypervariable intergenic sequence allows different isolates to be discriminated (Murthy et al. Reference Murthy, Dibbern and Campbell1992; Westenberger et al. Reference Westenberger, Sturm, Yanega, Podlipaev, Zeledon, Campbell and Maslov2004). Although this provides a useful barcode to identify different types of trypanosomatids, the sequences cannot be easily aligned. Therefore, we also attempted to sequence the highly conserved glycosomal GAPDH gene to reconstruct a phylogeny of Drosophila trypanosomatids, and examine how they are related to parasites from other hosts (Yurchenko et al. Reference Yurchenko, Lukes, Xu and Maslov2006). This work will give us a greater understanding of the parasites that infect this important model organism, and will identify Drosophila trypanosomatids that could be used to investigate host-parasite interactions.
MATERIALS AND METHODS
Field samples
We collected a range of different fly species using fruit baits from several European populations. In an urban population in Athens, Greece we collected Drosophila simulans and D. melanogaster over a 5-day period in October 2007 from an area ca. 300 m in diameter. Several related species from the obscura group were collected from woodland locations from around the UK in the summer of 2008. In addition a small collection of D. melanogaster was made in Braga, Portugal, which was used only for sequencing and is not included in prevalence estimates.
PCR and sequencing
To extract DNA, we homogenized single wild-caught flies, incubated the homogenate at 56°C for 1 h with Chelex 100TM ion exchange resin (Bio-Rad, Hercules, CA, USA) and proteinase-K, and then boiled the supernatant which was used directly for PCR. To check that the DNA extraction process had been successful, specific primers that amplify the ref(2)P gene in D. melanogaster and RpL32 in the obscura group were used. Any samples that did not amplify were discarded. Both D. melanogaster and D. simulans occur in the Greek population we sampled. Males were identified morphologically. The females were identified using PCR primers that amplify the drosomycin gene (dro-F 5′-CAG CCC TAA AGT ATG CCC TTC, dro-R 5′-AGC CAG GAA GAG GTA CAR GA) and yield products of 226 to 229 bp in D. melanogaster and 245 to 246 bp in D. simulans. The species in the obscura group were identified as D. obscura, D. subobscura, or D. helvetica using diagnostic PCR reactions that amplify the cytochrome b (Cyt-b) gene and the nuclear alcohol dehydrogenase (Adh) gene. A single specimen of D. tristis as well as 2 specimens of D. helvetica were identified by sequencing 852 bp of the Cyt-b gene, which were found to be identical to the published sequence from D. tristis (Accession number EF216284) and D. helvetica (Accession number EF216268) respectively.
To test flies for the presence of trypanosomatids, we used a diagnostic PCR that amplifies the trypanosomatid spliced leader sequence using the oligonucleotides M167, 5′-GGGAAGCTTCTGTACT(A/T)TATTGGTA, and M168, 5′-GGGAATTCAATA(A/T)AGTACAGAAACTG, that were described by (Westenberger et al. Reference Westenberger, Sturm, Yanega, Podlipaev, Zeledon, Campbell and Maslov2004). We sequenced the spliced leader sequence from a subsample of 21 DNA extractions from D. melanogaster, D. simulans, D. subobscura and D. helvetica (see Table 1). We used a cloning approach where direct sequencing of PCR products showed evidence of multiple sequences being present in the same fly. For 6 DNA samples, we cloned PCR fragments using the StrataClone PCR cloning kit (Stratagene, LaJolla, CA, USA) following the manufacturer's instructions and sequenced 4 clones per sample. Before sequencing, unused PCR primers and dNTPs were digested with exonuclease 1 and shrimp alkaline phosphatase. The PCR products were then sequenced directly using the PCR primers and Big Dye reagents (ABI) on an ABI capillary sequencer. Sequences were assembled using Sequencher 4.5 (Gene Codes Corporation) and chromatograms were inspected by eye to confirm the legitimacy of any differences between sequences. In total, we analysed 34 spliced leader sequences, including 19 variable sequences from potentially multiply infected individuals. Of these sequences, 27 represent a complete repeat unit, with the caveat that they include the overlapping primer binding sites in the conserved exon region (Table 1). We have included 7 sequences that are missing 50–51 bp of the intronic region downstream of the primer-binding site (Table 1). These sequences fully align with other complete sequences in our data set, including in the non-conserved intergenic region. The spliced leader sequences have been submitted to GenBank under the numbers HQ285325-HQ285358.
Table 1. Spliced leader repeat sequences

To reconstruct the parasite phylogeny, we sequenced the GAPDH genes from a subset of the infected flies following the protocol outlined above. To amplify the GAPDH gene we used the primers GAPDH_dir, 5′-AGAGGATCCATGGCTCCG(A/C)TCAAGGTTGGC-3′, and GAPDH_rev, 5′-AGAGGATCCTTACATCTTCGAGCTCGCG(C/G)(C/G)GTC-3′, that were described by Yurchenko et al. (Reference Yurchenko, Lukes, Xu and Maslov2006). The GAPDH sequences have been submitted to GenBank under the numbers HQ263664-HQ263666.
Data analysis
We obtained most of the available sequences of both genes from GenBank, ignoring multiple accessions, and aligned them with our sequences using ClustalW and corrected the alignment by eye. As the spliced leader sequences are highly divergent, only the most similar sequences can be reliably aligned, which means that while this gene is useful for typing different isolates, it does not produce reliable phylogenies of distantly related species (Westenberger et al. Reference Westenberger, Sturm, Yanega, Podlipaev, Zeledon, Campbell and Maslov2004). As our spliced leader sequences did not reliably align with sequences obtained from GenBank, we reconstructed neighbour-joining trees based on Jukes-Cantor genetic distances using the program Phylip (Felsenstein, Reference Felsenstein2005) for the Drosophila trypanosomatid samples based on contigs produced by Sequencher 4.5.
The phylogeny of the GAPDH amino acid sequences was reconstructed via a Bayesian approach using MrBayes (v. 3.1.2) (Huelsenbeck and Ronquist, Reference Huelsenbeck and Ronquist2001). We assumed a fixed rate model of protein evolution and reconstructed the phylogeny using a model jumping method. This method allows for different models of amino acid substitution to be used in the MCMC procedure, with all models contributing to the final result weighted according to their respective posterior probability. We ran 2 runs of 4 chains for 4 000 000 MCMC generations, sampling trees every 1000 generations. All trees were drawn using FigTree (http://tree.bio.ed.ac.uk/software/figtree/).
Additionally, we reconstructed the GAPDH phylogeny via the DNA sequences using both a Bayesian and a maximum-likelihood approach. Bayesian posterior support values are less conservative than maximum-likelihood bootstrap support, and so both the values can be used as an upper and lower support for nodes (Douady et al. Reference Douady, Delsuc, Boucher, Doolittle and Douzery2003). For the maximum likelihood trees, jModeltest (v. 0.1.1) (Guindon and Gascuel, Reference Guindon and Gascuel2003; Posada, Reference Posada2008) was used to estimate the model of sequence evolution and the analysis was run in PAUP (v. 4.0b10) (Swofford, Reference Swofford2003). A parsimony tree created from tree bisection and reconnection with a heuristic search was used as a starting tree for the maximum likelihood analysis. A GTR+G model with a gamma distribution of rate variation and no invariable sites was used. The maximum likelihood analysis used a heuristic search with a nearest neighbour interchange algorithm. The substitution rate parameters, shape of the gamma distribution and proportion of invariable sites used were those estimated by ModelTest. Support for the nodes was calculated by 1000 non-parametric bootstraps. Bayesian trees were created using the MrBayes program (v. 3.1.2) (Huelsenbeck and Ronquist, Reference Huelsenbeck and Ronquist2001). We used a general time-reversible model, with a gamma distribution and parameters estimated from the data during the analysis. As there is likely to be a considerable amount of noise from third codon positions between these divergent sequences, a site-specific rate model was used allowing each codon position to have its own rate. Two runs of 4 chains were run for 15 000 000 MCMC generations, with trees being sampled every 5000 generations. The DNA tree is presented as Supplementary material (online version only).
Differences in the proportion of infected flies were analysed in contingency tables using a Fisher Exact Test. In tables of more than 2 rows and 2 columns, significance was assessed by generating 100 000 random contingency tables with the same marginal values using a Monte Carlo procedure and taking the proportion with more extreme deviations as the probability (Lewontin and Felsenstein, Reference Lewontin and Felsenstein1965).
RESULTS
Prevalence
We tested 2129 wild-caught Drosophila for infection with trypanosomatids using a diagnostic PCR reaction that amplifies the spliced leader RNA gene. All 5 identified species that we collected – D. melanogaster, D. simulans, D. obscura, D. subobscura and D. helvetica – were infected (Table 2). Note that we did not further determine the species of uninfected flies from the obscura group by sequencing. The prevalence of infection varied significantly among these species (Fisher Exact Test: P=0·02), but was always below 5%. This variation was not entirely caused by samples coming from different geographical regions, as the prevalence differed between the 2 species from Greece (D. melanogaster and D. simulans; Fisher Exact Test: P=0·02) but not the 2 from the UK (D. obscura and D. subobscura; Fisher Exact Test: P=0·36). Multiple spliced leader sequences from a single fly were detected in 28·5% of samples, suggesting that these flies may have been infected by 2 trypansomatids with distinct spliced leader RNA genotypes. Alternatively, this may indicate microheterogeneity of spliced leader repeat units, which has been reported in both Leishmania major and Trypanosoma cruzi (Thomas et al. Reference Thomas, Westenberger, Campbell and Sturm2005).
Table 2. The prevalence of trypanosomatids in four species of Drosophila

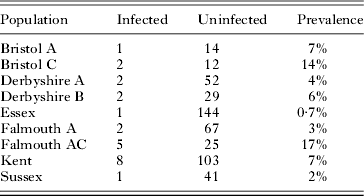
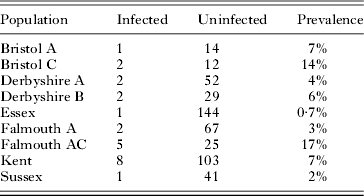
Drosophila obscura and D. subobscura were collected from 14 sites scattered across the UK. In D. obscura, there was significant geographical variation in prevalence (Table 3; Fisher Exact Test: P=0·03). For example, in the 2 populations with the largest sample size the prevalence ranged from 0·7% to 7%. In D. subobscura, where we had smaller samples, there was no significant geographical variation in prevalence (data not shown; Fisher Exact Test: P=0·08).
Table 3. The prevalence of trypanosomatids in different populations of Drosophila obscura from across the UK
Barcoding trypanosomatid samples
To distinguish different trypanosomatids we sequenced the spliced leader sequence. The spliced leader RNA is encoded in the genome in many copies arranged in a tandem array. The sequence of each repeat consists of a highly conserved exon, a variable intron and a hypervariable intergenic sequence (Murthy et al. Reference Murthy, Dibbern and Campbell1992). It has been found that a 20 bp sequence of the exon can be used to define groups of related species, and it is therefore a useful ‘barcode’ that can be used to identify trypanosomatids to a level akin to a genus (Westenberger et al. Reference Westenberger, Sturm, Yanega, Podlipaev, Zeledon, Campbell and Maslov2004; Maslov et al. Reference Maslov, Westenberger, Xu, Campbell and Sturm2007).
We sequenced this 20 bp region and found that it was identical in 33 of 34 individual sequences (AACTAACGCTATTATTGTTA), suggesting that most of these trypanosomatids are closely related. The hosts consisted of 14 Greek D. melanogaster and 2 Greek D. simulans, 2 UK D. subobscura and 2 UK D. helvetica. Barcoding assigns these trypanosomatid samples to a group consisting of Sergeia podlipaevi, several Herpetomonas species, one species of Leptomonas and several unidentified Trypanosomatidae isolated from Heteroptera in Egypt and Southwest China. The only other sequence that was identified was from a single D. subobscura from the UK, which was identical to that found in the ‘SE clade’ (AACTAACGCTATATAAGTAT, Maslov et al. Reference Maslov, Westenberger, Xu, Campbell and Sturm2007). This highly specious group contains numerous isolates from many species of insects, and includes the genera Crithidia and Leptomonas, which contain a number of insect parasites.
Although the exon is useful to identify major groups of trypanosomatids, it evolves too slowly to distinguish more closely related species or strains. To do this we examined 1 spliced leader RNA repeat unit. Examining the sequences, it was clear that they fell into 5 major sequence groups (Fig. 1). Groups A-D all share the exon sequence AACTAACGCTATTATTGTTA and group E has the sequence AACTAACGCTATATAAGTAT. The sequence groups show variation in repeat length (see Table 1), which is caused by indels in the highly variable intergenic region. Additionally, groups D and E show small indels in the intronic region (a 1 bp insertion and a 5 bp deletion respectively). The pair-wise genetic similarity between sequences within each group is very high, with an average of 95·4% in group A, 94·7% in group B and 99·8% in group C, and both the intergenic and intronic regions could be aligned. The maximum pair-wise genetic distance in these groups is 12·6%, 7·7% and 0·2% respectively. However, between the groups there is a very high level of divergence, and, although some stretches of homologous sequence are clearly visible in the intronic region, reliable sequence alignments across the repeat unit were impossible. Therefore there are distinct species or strains of trypanosomatids that infect Drosophila. We refrained from further phylogenetic analyses of the spliced leader sequence because these methods rely on perfect alignments (Ogden and Rosenberg, Reference Ogden and Rosenberg2006), and reliable alignments were not possible.

Fig. 1. Neighbour-joining trees of the spliced leader sequences illustrating genetic diversity within sequence groups. The taxa are labelled according to the species of Drosophila the trypanosomatids were found in, following the nomenclature in Table 1. The sequences form 5 groups (A–E), within which the genetic distance between sequences is 13% or less. The sequences in the different groups are too divergent to reliably align (except for the conserved region).
To examine how the strains are distributed across our different fly species, we made separate sequence alignments of each sequence group and reconstructed neighbour-joining trees of these sequences (Fig. 1). It is clear that there are strong effects of host species in which genotypes of trypanosomatids are found. However, there are no significant differences between the groups that are found in closely related species collected from the same location and habitat – Greek D. simulans and D. melanogaster, or members of the obscura group from the UK – suggesting that there is transmission of parasites between these species.
Phylogeny
The rapid evolution of the spliced leader RNA sequence means that it cannot be used to infer the phylogeny of distantly related strains of trypanosomatids, so instead we attempted to amplify the GAPDH gene (glyceraldehyde-3-phosphate dehydrogenase), which is more conserved (Yurchenko et al. Reference Yurchenko, Lukes, Xu and Maslov2006). Unfortunately, this proved to be difficult, and we only obtained sequences from 3 trypanosomatid samples found in D. tristis and D. obscura from the UK, and D. melanogaster from Portugal, from which, in turn, we failed to obtain the spliced leader sequence. Similar difficulties in sequencing both the spliced leader and GAPDH gene have been reported before (i.e. Votypka et al. Reference Votypka, Maslov, Yurchenko, Jirku, Kment, Lun and Lukes2010; Maslov et al. Reference Maslov, Yurchenko, Jirku and Lukes2010) and Maslov et al. (Reference Maslov, Yurchenko, Jirku and Lukes2010) have pointed out that additional phylogenetic markers will need to be analysed in order to resolve the trypanosomatid phylogeny. As the sequences were highly divergent, we reconstructed phylogenies using protein sequences as well as DNA sequences. The D. tristis and D. obscura trypanosomatid samples form a monophyletic group that is distant from the sample found in D. melanogaster (Fig. 2 and S1-Online version only). The sequences are most closely allied to other monoxenous species that infect insects. The closest relatives of the D. melanogaster trypanosomatid sample are Herpetomonas muscarum, H. megaseliae and ‘isolate 37EC’, which naturally infect house flies, scuttle flies and a heteropteran bug (Yurchenko et al. Reference Yurchenko, Lukes, Jirku and Maslov2009). There is only weak support for the relationships of the D. tristis and D. obscura trypanosomatid samples to other species, but the most likely relative is a parasite of mosquitoes, Blastocrithidia culicis.

Fig. 2. Phylogeny of the GAPDH gene reconstructed from protein sequences. The tree was reconstructed using the Bayesian method and node labels show the posterior support for the clade.
DISCUSSION
We have found that trypanosomatids are common parasites of Drosophila, and have for the first time characterized the diversity of these parasites in natural populations. The sequences from the spliced leader sequence show that the large majority of trypanosomatids circulating in European Drosophila populations are genetically distinct from those found in other large surveys of trypanosomatids as it was impossible to align the sequences. Within this group there are several distinct genotypes that can be identified from the more variable regions of the spliced leader sequence, and it is possible that these are different species of the parasites. How these different strains relate to other trypanosomatids is still uncertain, as we had difficulty in sequencing more conserved genes from the parasites, failing to obtain any GAPDH sequences from the samples for which we sequenced the spliced leader sequence and vice versa. However, the sequences that we did obtain show that they are most closely related to other monoxenous species that only infect insects (as opposed to the dixenous species of the two genera Trypanosoma and Leishmania that infect vertebrates), and belong to at least 2 different lineages on the trypanosomatid phylogeny.
The PCR approach that we have used may detect very low numbers of parasites. It is possible that some of the trypanosomatids detected may have been picked up during feeding in the wild, without having replicated significantly in the fly. However, this is unlikely as all the flies were maintained on agar or fly medium for several days before the PCR assay. Additionally, it is known that naturally occurring trypanosomatid infections do establish in Drosophila (e.g. Ebbert et al. Reference Ebbert, Burkholder and Marlowe2001, Reference Ebbert, Marlowe and Burkholder2003). Furthermore, the infection in the Portuguese specimen was detected by microscopy (Ferreira, unpublished data); this sample showed morphological similarity to Leptomonas drosophilae as described by Chatton and Leger (Reference Chatton and Leger1911). Further work is required to establish whether all the organisms that we have detected are true parasites of these species of Drosophila.
Drosophila is widely used as a model species to study both invertebrate immune systems and the evolution of hosts and parasites. Trypanosomatids are a potential new system for these studies, as they represent one of the few natural parasites of Drosophila that we know of and many species can be cultured in vitro. They may be naturally co-evolving with Drosophila, potentially leading to flies evolving specific immune responses to counter these infections, and in turn parasites having been selected to evade Drosophila immune responses. Much of the research into the Drosophila immune systems has focussed on systemic immune responses to bacteria and fungi. It is therefore also interesting to find that protists that infect the gut are common in fly populations, and studying the immune response to these parasites promises to reveal new aspects of the invertebrate immune response. As Drosophila is a model system particularly amenable to molecular and immunological studies, this avenue of research also has the potential to yield new methods of fighting monoxenous trypanosomatid infections in economically important insects such as honeybees and bumblebees as well as in dixenous infections in which insects vector vertebrate parasites. Important ecological questions also remain unanswered. We found that different genotypes of the parasites are found in different host species. It is currently unclear whether this host specificity is the result of parasites being physiologically adapted to certain host species, or if it instead emerges simply because related species share related habitats or were collected from the same sites. In conclusion, trypanosomatids represent an exciting new opportunity for evolutionary and immunological research in Drosophila.
ACKNOWLEDGEMENTS
We would like to thank Claire Webster and Mitchel Chewi for assistance in the laboratory and Natasa Fytrou and Heather Cagney for help with fieldwork.
FINANCIAL SUPPORT
This work was supported by the Leverhulme Trust (F.M.J., F/00 158/BJ). F.M.J was supported by a Royal Society University Research Fellowship and B.L. by a BBSRC Studentship.