


TRYPANOSOMES COMPARTMENTALIZE GLYCOLYSIS AND OTHER PATHWAYS WITHIN GLYCOSOMES
A remarkable feature of protists belonging to the clade Kinetoplastea is the compartmentalization of the major part of the glycolytic pathway and other important pathways involved in catabolism and anabolism inside peroxisome-related organelles called glycosomes (reviewed by Gualdrón-López et al. Reference Gualdrón-López, Brennand, Hannaert, Quiñones, Cáceres, Bringaud, Concepción and Michels2012a). This sequestering of glycolytic enzymes was first detected, in 1977, for bloodstream-form Trypanosoma brucei (Opperdoes and Borst, Reference Opperdoes and Borst1977), but subsequently also for Crithidia fasciculata and Trypanosoma cruzi, Leishmania spp. and other Trypanosomatida (Taylor et al. Reference Taylor, Berghausen, Heyworth, Messenger, Rees and Gutteridge1980; Hart and Opperdoes, Reference Hart and Opperdoes1984; Sánchez-Moreno et al. Reference Sánchez-Moreno, Lasztity, Coppens and Opperdoes1992) and Parabodina (Opperdoes et al. Reference Opperdoes, Nohynkova, Van Schaftingen, Lambeir, Veenhuis and Van Roy1988; Ardelli et al. Reference Ardelli, Witt and Woo2000).
In all trypanosomatids studied, the conversion of glucose to 3-phosphoglycerate, or in some cases to 1,3-bisphosphoglycerate, occurs within the glycosomes, by the 7 or 6 successive glycolytic enzymes from hexokinase (HXK) to phosphoglycerate kinase (PGK) or glyceraldehyde-3-phosphate dehydrogenase (GAPDH), respectively. The 3 or 4 last enzymes of the pathway, from phosphoglycerate mutase (PGAM) – or PGK – to pyruvate kinase (PYK) are present in the cytosol. This organization represents the situation in the proliferating long-slender bloodstream-forms of T. brucei, that convert glucose, taken up from the blood of the mammalian host, into pyruvate that is excreted again in the bloodstream. Glycolysis is the only ATP-providing process in these parasites when they multiply in mammals including humans. The pathway is organized in such a way that no net change in the ATP and ADP concentrations occurs inside the organelles (Fig. 1). Per molecule of glucose consumed, 2 molecules of ATP are used in the first part of the glycolytic pathway, in the reactions catalysed by HXK and phosphofructokinase (PFK), whereas 2 ATP molecules are produced downstream in the pathway by the activity of PGK. The net production of ATP, 2 molecules per glucose, occurs in the PYK reaction in the cytosol.

Fig. 1. The glycolytic pathway in bloodstream-form Trypanosoma brucei. Glucose is the substrate, the end products pyruvate and glycerol are boxed. The enzymes catalysing the reactions are indicated by numbers: 1, hexokinase; 2, glucose-6-phosphate isomerase; 3, phosphofructokinase; 4, aldolase; 5, triosephosphate isomerase; 6, glyceraldehyde-3-phosphate dehydrogenase; 7, phosphoglycerate kinase; 8, NAD-dependent glycerol-3-phosphate dehydrogenase; 9, glycerol kinase; 10, phosphoglycerate mutase; 11, enolase; 12, pyruvate kinase; 13, FAD-dependent glycerol-3-phosphate dehydrogenase; 14, ubiquinone; 15, SHAM-sensitive alternative oxidase. Circles in the glycosomal membrane indicate translocation processes of which the proteins involved have not yet been identified. Abbreviations: 1,3BPGA, 1,3-bisphosphoglycerate; DHAP, dihydroxyacetone phosphate; e−, electrons; F6P, fructose 6-phosphate; FBP, fructose 1,6-bisphosphate; GAP, glyceraldehyde 3-phosphate; G3P, glycerol 3-phosphate; G6P, glucose 6-phosphate; PEP, phosphoenolpyruvate; 2PGA, 2-phosphoglycerate; 3PGA, 3-phosphoglycerate. The dashed arrow for the glycerol kinase reaction indicates that this reaction, in the direction of glycerol formation with concomitant ATP synthesis, can only occur under anaerobic condition (the thermodynamically favoured reaction catalysed by this enzyme is the ATP-dependent phosphorylation of glycerol).
In glycolysis, the substrate is oxidized with the concomitant formation of NADH in the GAPDH-catalysed reaction inside the organelle. However, also no net change occurs in the intraglycosomal NAD/NADH ratio during glycolysis, because the electrons are transferred from the glycosomal matrix to the mitochondrion via a shuttle mechanism by which NADH is re-oxidized by a glycosomal glycerol-3-phosphate dehydrogenase (G3PDH). In this step, dihydroxyacetone phosphate (DHAP) is reduced to glycerol 3-phosphate (G3P). The G3P is then transported by a putative G3P/DHAP antiporter to the cytosol, to be oxidized by a mitochondrial glycerol-3-phosphate oxidase (GPO) system to DHAP that enters the glycosome in exchange for the G3P. The GPO exists of a FAD-dependent G3PDH associated with the outer face of the mitochondrial inner membrane, ubiquinone and an alternative oxidase (AO). The mitochondrial GPO activity is not coupled to ATP synthesis. In contrast to the other life-cycle stages, and the different life-cycle stages of other parasitic trypanosomatids, the mitochondrial metabolic repertoire is severely repressed in bloodstream-form T. brucei. It has no tricarboxylic acid (TCA) cycle, cytochromes-containing respiratory chain and capacity for oxidative phosphorylation.
Under anaerobic conditions or when the AO is inhibited by salicylhydroxamic acid (SHAM), the glucose is not converted almost exclusively into pyruvate, as under aerobic conditions, but in equimolar amounts of pyruvate and glycerol. In this case intraglycosomal G3P is dephosphorylated. This does not occur by a glycerol-3-phosphatase, but by the reverse action of a glycerol kinase (GK), located in the glycosomal matrix, with the concomitant synthesis of ATP. Although the equilibrium of the GK-catalysed reaction strongly favours the ATP-dependent phosphorylation of glycerol, it was hypothesized that anaerobiosis causes an intraglycosomal accumulation of G3P and low ATP/ADP ratio that enables the reversal of the GK reaction (Opperdoes and Borst, Reference Opperdoes and Borst1977). Indeed, this has been confirmed by computer simulation with a kinetic model of bloodstream-form glycolysis, as well as experimentally (Hammond et al. Reference Hammond, Aman and Wang1985; Bakker et al. Reference Bakker, Michels, Opperdoes and Westerhoff1997; Haanstra et al. Reference Haanstra, Van Tuijl, Kessler, Reijnders, Michels, Westerhoff, Parsons and Bakker2008). The peculiar organization of the pathway allows glycolysis still to occur under anaerobic conditions, but then with the conversion of the 2 triosephosphates produced by the aldolase reaction following separate routes: glyceraldehyde 3-phosphate to pyruvate, and DHAP to glycerol (Fig. 1). Two molecules of ATP remain to be synthesized inside the organelle, one by PGK and one by GK, balancing the ATP consumption by HXK and PFK, but the net production of ATP, by PYK in the cytosol, is halved.
Bloodstream-form trypanosomes can be maintained for several hours under anaerobic conditions or in the presence of SHAM, but they do not proliferate and eventually die (Helfert et al. Reference Helfert, Estévez, Bakker, Michels and Clayton2001). The possibility of anaerobic glycolysis, with reduced ATP synthesis, could be considered as a means for trypanosomes to survive during periods that low oxygen tension is experienced. Additionally, partial depletion of various glycolytic enzymes leading to a reduced glycolytic flux has shown that decreasing the flux by approximately 50% is sufficient to halt growth and leads to death of the parasites (Haanstra et al. Reference Haanstra, Kerkhoven, Van Tuijl, Blits, Wurst, Van Nuland, Albert, Michels, Bouwman, Clayton, Westerhoff and Bakker2011).
The importance of glycolysis for bloodstream-form trypanosomes, as their only process for ATP supply, is underscored by the high glycolytic flux and the fact that up to 90% of the glycosomes’ protein content is made up of glycolytic enzyme (Opperdoes et al. Reference Opperdoes, Baudhuin, Coppens, De Roe, Edwards, Weijers and Misset1984; Bakker et al. Reference Bakker, Westerhoff and Michels1995; Haanstra et al. Reference Haanstra, Van Tuijl, Kessler, Reijnders, Michels, Westerhoff, Parsons and Bakker2008; Gualdrón-López et al. Reference Gualdrón-López, Brennand, Hannaert, Quiñones, Cáceres, Bringaud, Concepción and Michels2012a). The enzymes are packed into the organelles, which constitute about 5% of the total cell volume, at the relatively high protein concentration of 150 mg/ml (Misset et al. Reference Misset, Bos and Opperdoes1986). In contrast, in insect-stage, procyclic T. brucei, and in different life-cycle stages of other trypanosomatid parasites studied the glycolytic flux is much lower. These cells have a de-repressed mitochondrial metabolism involving active TCA cycle enzymes, a cytochromes-containing respiratory chain and an oxidative phosphorylation machinery. Other substrates for ATP production, such as fatty acids and amino acids may then be used, dependent on the host, necessitating a gluconeogenic rather than a glycolytic flux in order to produce sugar-phosphates for the production of glycoconjugates, oligo- and polysaccharides. Gluconeogenesis also occurs in the glycosomes, as well as other processes – or parts of them – such as fatty-acid β-oxidation, purine salvage, and the biosynthesis of pyrimidines, sterols, ether lipids and sugar nucleotides. Several of these pathways are up-regulated compared to bloodstream-form T. brucei, whereas the expression of glycolytic enzymes is down-regulated to 40–50% in procyclic T. brucei and possibly even lower in some other trypanosomatids [reviewed by Gualdrón-López et al. (Reference Gualdrón-López, Brennand, Hannaert, Quiñones, Cáceres, Bringaud, Concepción and Michels2012a, Reference Gualdrón-López, Michels, Quiñones, Cáceres, Avilán, Concepción, Jäger, Koch and Flohéc)]. When procyclic T. brucei and other trypanosomatids catabolize sugars, the glycosomal glycolytic pathway is extended to a succinate fermentation pathway.
When T. brucei parasites live in the blood of humans, they cause sleeping sickness that will have a fatal outcome if not treated. No adequate treatment is available; the currently available drugs are toxic, not very efficacious, difficult to administer and/or resistance against them has developed. There is an urgent need to develop new and safe compounds for anti-parasitic chemotherapy. Since these trypanosomes are completely dependent on glycolysis for their ATP supply, glycolytic enzymes have been indicated as potential drug targets. The notion of trypanosomal glycolytic enzymes as promising targets was reinforced by the finding of their compartmentalization inside glycosomes. It was reasoned that the unique organization, combined with the distant evolutionary relationship with their human glycolytic enzymes must have endowed the enzymes of the parasites with unique structural and functional properties that could be exploited for drug discovery (Opperdoes, Reference Opperdoes1985). This hypothesis has been amply validated in subsequent research [reviewed by (Opperdoes and Michels, Reference Opperdoes and Michels2001; Verlinde et al. Reference Verlinde, Hannaert, Blonski, Willson, Périé, Fothergill-Gilmore, Opperdoes, Gelb, Hol and Michels2001; Gualdrón-López et al. Reference Gualdrón-López, Michels, Quiñones, Cáceres, Avilán, Concepción, Jäger, Koch and Flohé2012c)].
Glycosomes are bounded by a single phospholipid membrane. Several observations provide support for the notion that this membrane forms a permeability barrier. First, as mentioned above, under anaerobic conditions, glycerol can be formed with concomitant ATP synthesis owing to the accumulation of G3P and a lowered ATP/ADP ratio inside the glycosomes, allowing the reversal of the GK reaction. A rapid equilibrium of G3P and adenine nucleotides across the membrane would prevent this reversal. Secondly, an apparent low equilibrium of glycolytic intermediates across the membrane was observed upon addition of a pulse of radioactively labelled glucose to trypanosomes. A small (20–30%) pool of intermediates, as well as the end-product pyruvate were very rapidly labelled, whereas the label appeared only 60 times slower in a large (70–80%) pool of intermediates. The small pool was interpreted as the metabolites present inside the glycosomes, and the large pool as those in the cytosol (Visser et al. Reference Visser, Opperdoes and Borst1981). Furthermore, computer simulation of glycolysis using the earlier mentioned kinetic model indicated the essentiality of a defined pool of phosphorylated glycolytic intermediates within the glycosome, separate from the cytosol, to enable a functional glycolytic flux (Bakker et al. Reference Bakker, Michels, Opperdoes and Westerhoff1997). Moreover, several of the glycolytic glycosomal enzymes possess peculiar functional properties that restrict their proper functioning to within the glycosomal boundaries. For example, trypanosomatid HXK and PFK lack the activity regulation mechanisms found in their counterparts of other organisms that serve to prevent uncontrolled enzyme activity that might result from the ‘turbo design’ of the pathway (Bakker et al. Reference Bakker, Mensonides, Teusink, Van Hoek, Michels and Westerhoff2000). These mechanisms prevent overstimulation by the net ATP synthesized in glycolysis, which could lead to excessive accumulation of the downstream intermediates. However, the sequestering of the first 7 enzymes of the pathway within the organelles creates an environment in which ATP consumption and production are balanced, thus preventing HXK and PFK activation by the net ATP produced by PYK in the cytosol. Indeed, by computer simulation and experimentally, it has been shown that relocation of the enzymes to the cytosol results in huge accumulation of intermediates in the cell that may be the cause of, or contribute to the associated death of the trypanosomes (Bakker et al. Reference Bakker, Mensonides, Teusink, Van Hoek, Michels and Westerhoff2000; Haanstra et al. Reference Haanstra, Van Tuijl, Kessler, Reijnders, Michels, Westerhoff, Parsons and Bakker2008). This suggests that the glycosomal membrane prevents the matrix enzymes from directly sensing the cytosolic ATP.
GLYCOSOMES BELONG TO THE ORGANELLE FAMILY OF PEROXISOMES
Many properties of glycosomes may be best understood through the relationship of these organelles with peroxisomes. The organelles belonging to the peroxisome family can be very diverse in morphology. They may differ greatly in size, can form round or elongated vesicles and even have lamellar appearances or form a dynamic peroxisomal network. However, they all have in common a single boundary phospholipid membrane, often with a dense protein matrix. Furthermore, the organelles can be very different in the enzymes they contain. In most organisms peroxisomes contain enzymes involved in peroxide metabolism, often H2O2-producing oxidases and almost universally H2O2-detoxifying enzymes such as catalase and peroxidase. In addition, in many peroxisomes enzymes of ether-lipid biosynthesis, fatty-acid oxidation and the oxidative branch of the pentosephosphate pathway (PPP) are found. Glycosomes of trypanosomatids do not contain H2O2-producing oxidases and catalase, but the latter enzyme has been found in the glycosomal cell fraction of the bodonid Trypanoplasma borreli (Opperdoes et al. Reference Opperdoes, Nohynkova, Van Schaftingen, Lambeir, Veenhuis and Van Roy1988). In addition, glycosomes have acquired other enzymes important for dealing with reactive oxygen such as those involved in ascorbate synthesis (reviewed by Gualdrón-López et al. Reference Gualdrón-López, Brennand, Hannaert, Quiñones, Cáceres, Bringaud, Concepción and Michels2012a, Reference Gualdrón-López, Michels, Quiñones, Cáceres, Avilán, Concepción, Jäger, Koch and Flohéc).
Most importantly, all members of the peroxisome family, including glycosomes, seem to follow a similar route of biogenesis (reviewed by Moyersoen et al. Reference Moyersoen, Choe, Fan, Hol and Michels2004; Galland and Michels, Reference Galland and Michels2010). For various organisms – not yet for trypanosomatids – it has been shown that new organelles are formed by growth and division of existing peroxisomes. Peroxisomes and glycosomes do not contain a genome; all their proteins are encoded in the nucleus. New proteins are post-translationally imported from the cytosol into the matrix or inserted into the membrane, and lipids are obtained from the endoplasmic reticulum (ER) by non-vesicular transport. However, recent research on peroxisomes proved that the organelles can also be formed de novo, whereby lipids and specific membrane proteins are recruited via membrane vesicles from distinct regions of the ER. The different stadia of the biogenesis of these organelles are carried out by specific proteins called peroxins (acronym PEX). Currently a combined, not redundant set of 33 peroxins are known from different organisms. A considerable number of these peroxins have been found as homologous proteins in organisms as diverse as mammals, plants, yeasts and protists like trypanosomatids, although with little sequence conservation. Some of the peroxins are homologous to proteins involved in the ER-associated protein degradation (ERAD) system (Schliebs et al. Reference Schliebs, Girzalsky and Erdmann2010). Together, these data indicate that all members of the peroxisome family are derived from a common ancestral organelle that was derived from the ER. The presence of peroxisomes in representatives of all eukaryotic supergroups indicates that this ancestral origin has taken place already in the last eukaryotic common ancestor (often referred to as LECA). Glycosomes seem to have originated in a common ancestor of the Kinetoplastea and Diplonemida, from a peroxisome that acquired the glycolytic pathway. How this may have happened, and what has been the selective advantage of the acquisition of the glycosome have been discussed in detail elsewhere (Gualdrón-López et al. Reference Gualdrón-López, Brennand, Hannaert, Quiñones, Cáceres, Bringaud, Concepción and Michels2012a). The corollary of this common origin is that peculiar properties of peroxisomes may also be relevant for the understanding of glycosomes, as will be illustrated below.
GLYCOSOME BIOGENESIS; PROTEIN TRANSLOCATION ACROSS THE GLYCOSOMAL MEMBRANE
Biogenesis of peroxisomes and glycosomes involves several processes such as the matrix-protein import, acquisition of membrane lipids and insertion of membrane proteins. As mentioned above, it has been shown for peroxisomes that they can be formed by growth of existing organelles, in which case daughter organelles are produced from mother peroxisomes by division or budding. Here we will focus on how matrix proteins are translocated across the membrane from the cytosol into the peroxisomes/glycosomes. Currently it is not known whether this import occurs in all existing peroxisomes or only in a fraction that is import-competent, and/or newly formed pre-peroxisomal vesicles derived from the ER.
Proteins are imported into these organelles post-translationally, usually without undergoing any form of processing. Import occurs without a requirement for unfolding (Häusler et al. Reference Häusler, Stierhof, Wirtz and Clayton1996); even oligomeric proteins may be imported (McNew and Goodman, Reference McNew and Goodman1994; Titorenko et al. Reference Titorenko, Nicaud, Wang, Chan and Rachubinski2002; Léon et al. Reference Léon, Goodman and Subramani2006), although this is not a general feature of peroxisomal matrix proteins (Faber et al. Reference Faber, Van Dijk, Keizer-Gunnink, Koek, Van der Klei and Veenhuis2002). The proteins to be imported do generally not have cleavable targeting signals. The proteins are routed to the organelles by their so-called peroxisomal-targeting signals (PTS) that are similar for all members of the organelle family, including glycosomes. Two amino-acid consensus motifs are known to function as PTS. The most common one is PTS1, the C-terminal sequence SKL, or a comparable tripeptide with at each position a different amino acid with similar physicochemical properties (Brocard and Hartig, Reference Brocard and Hartig2006). The alternative PTS2 is a nonapeptide located close to the N-terminus with the consensus (R/K), (L/V/I), X5, (H/Q), (L/A) where X is any residue (Lazarow, Reference Lazarow2006). Targeting may also be achieved by a polypeptide internal sequence or I-PTS. However, internal signals of different proteins do not have a consensus motif and the manner by which they function and the proteins that they use to enter the organelles have not been unambiguously established. At least in some cases this may involve piggy-back transport on a PTS1 or PTS2 containing protein (Van der Klei and Veenhuis, Reference Van der Klei and Veenhuis2006; Galland et al. Reference Galland, de Walque, Voncken, Verlinde and Michels2010).
PTS proteins are recognized in the cytosol by receptors which bring them to the surface of the peroxisomes/glycosomes: PTS1 proteins by PEX5, PTS2 proteins by PEX7 (Fig. 2). In the case of PTS2-protein/PEX7, at least in yeasts and fungi, one or more co-receptors may be involved: PEX18, PEX20 and PEX21 (Schliebs and Kunau, Reference Schliebs and Kunau2006; Galland and Michels, Reference Galland and Michels2010). These latter peroxins are required to form an import-competent complex with PEX7 together with the PTS2 protein. In contrast, in mammals, plants and most likely also in trypanosomes, it is PEX5 that fulfils the role of PEX7 co-receptor; the cargo-loaded PEX7 binds to PEX5 (Schliebs and Kunau, Reference Schliebs and Kunau2006; Galland et al. Reference Galland, Demeure, Hannaert, Verplaetse, Van Der Smissen, Courtoy and Michels2007) and forms a ternary complex [reviewed by (Galland and Michels, Reference Galland and Michels2010)].

Fig. 2. Diagram of the import of glycosomal matrix proteins in Trypanosoma brucei. Matrix protein import is achieved by a process which involves cyclic receptors, here depicted for PEX5, the receptor of matrix proteins with a PTS1 motif. Six distinct steps can conceptually be understood: (I) binding of proteins with a PTS1 to PEX5 (and PTS2 to PEX7, plus PEX7-PTS2-proteins to PEX5). (II) Docking of the receptor-cargo complex to the docking complex (PEX13.1, PEX13.2, PEX14) in the glycosomal membrane, followed by the insertion of PEX5 into the membrane, forming in conjunction with PEX14 a transient pore. (III) Release of PTS proteins, via the transient pore, into the matrix. (IV) Ubiquitination of the membrane-inserted PEX5 by the coordinated action of a cytosolic ubiquitin-conjugating enzyme PEX4, anchored to the membrane by PEX22, and an ubiquitin-ligating enzyme of the RING-finger complex, which comprises PEX2, PEX10 and PEX12. (V) Retrieval of the ubiquitinated PEX5 from the membrane by the cytosolic AAA+-ATPase complex including PEX1 and PEX6, recruited to the membrane. (VI) Deubiquitination of PEX5, rendering the receptor available to mediate new rounds of PTS-protein import. All peroxins identified in T. brucei are indicated in this scheme by the numbers attributed previously to their S. cerevisiae homologues. Question marks indicate yeast peroxins of which the trypanosomatid equivalents have not (yet) been identified.
The import of the PTS proteins occurs in a process by which the receptors cycle between the cytosol and organelles and can perform repeated rounds of protein import (Fig. 2). The overall pattern of this process has been solved for yeasts, notably S. cerevisiae (reviewed in Girzalsky et al. Reference Girzalsky, Platta and Erdmann2009; Williams and Stanley, Reference Williams and Stanley2010; Rucktäschel et al. Reference Rucktäschel, Girzalsky and Erdmann2011) – without yet revealing many mechanistic details – and at least in parts observed to be similar in other organisms including trypanosomes (Galland and Michels Reference Galland, de Walque, Voncken, Verlinde and Michels2010; M.G.L. and P.M., unpublished observations). The next step in the cycle, after the binding of the PTS-proteins to the receptors, is the interaction of the cargo-loaded receptor with a docking complex associated with the peroxisomal/glycosomal membrane. In all organisms studied, the docking complex comprises at least 2 proteins, the integral membrane protein PEX13 and the membrane associated peroxin PEX14, but may contain additional proteins, such as PEX17 in S. cerevisiae. In trypanosomes two very different isoforms of PEX13 (PEX13.1 and PEX13.2), both essential, have been detected (Verplaetse et al. Reference Verplaetse, Rigden and Michels2009; Brennand et al. Reference Brennand, Rigden and Michels2012).
The step after docking, which is the cargo translocation through the peroxisomal membrane, has been a mystery for many years. But recently, Meinecke et al. (Reference Meinecke, Cizmowski, Schliebs, Krüger, Beck, Wagner and Erdmann2010) identified in yeast peroxisomal membranes large complexes (600–800 kDa) comprising PEX5 and PEX14. Electrophysiological analysis of these complexes in liposomes indicated that they form pores with low but variable conductance states and ion selectivity; the main channel had an estimated diameter of 0·6 nm. Based on the assumption that the pores were transient, subject to continuous disassembly, the complexes were purified from yeast mutants not having PEX8 (see below) and the PEX7 co-receptors. Indeed a more stable pore with a diameter of 3·8 nm could be obtained. This pore appeared to be gated: after its docking, the cargo-receptor complex causes the widening of the channel to a size of over 9 nm, sufficient to allow the passage of large and oligomerized proteins. Indeed, previous structural analysis of PEX5 had suggested that this peroxin may be able to undergo major conformational changes and consequently occurs both as a soluble protein and an oligomeric membrane complex (Erdmann and Schliebs, Reference Erdmann and Schliebs2005; Stanley and Willmanns, Reference Stanley and Wilmanns2006). The studies by Meinecke et al. (Reference Meinecke, Cizmowski, Schliebs, Krüger, Beck, Wagner and Erdmann2010) suggest that the soluble cargo-loaded PEX5, after docking, may associate with the pore complex and either replaces a cargo-empty PEX5 or inserts itself as an extra component, causing the change in the size of the channel and allowing the cargo to be translocated.
The docking and pore complex or importomer also associates, via PEX8, with another complex, responsible for the retrieval of the receptor after it has released its cargo into the lumen of the organelle. The retrieval involves the ubiquitination of PEX5, and possibly also the peroxins involved in PTS2 protein delivery, such as PEX7 and, in yeasts and fungi, its co-receptor(s) [reviewed by (Rucktäschel et al. Reference Rucktäschel, Girzalsky and Erdmann2011)]. Whereas PEX5 has to be retrieved from the membrane, PEX7 seems to be delivered within the lumen with its cargo and has to be exported again from it (Nair et al. Reference Nair, Purdue and Lazarow2004). Nevertheless, no mechanistic details concerning PEX7 retrieval from the luminal side of the peroxisomes have been proposed. The retrieval of PEX5 from the membrane involves the action of several subcomplexes: (i) a complex of 2 predominantly cytosolic ATPases of the AAA+-protein family (ATPases associated with various cellular activities), PEX1 and PEX6, that transiently binds, via PEX6, to the membrane through PEX15 in yeast and PEX26 in mammalian cells; (ii) a complex of 3 RING-finger proteins (PEX2, PEX10 and PEX12), present in the peroxisomal membrane, and (iii) a complex formed, in yeast and plants, by the a cytosolic PEX4 and its membrane anchor PEX22, which plays a role in the process by the ubiquitination of PEX5 to signal that it is to be retrieved. The RING-finger proteins have E3-ubiquitin ligase activity whereas PEX4 belongs to the family of E2 ubiquitin-conjugating enzymes. In mammals the E2 activity in the process is exerted by the isoforms UbcH5a/b/c that are not associated with the membrane.
The retrieval of the receptor may involve the conjugation of only a single ubiquitin moiety or poly-ubiquitination. The latter modification seems rather to serve in quality control and leads to disposal of dysfunctional receptors in proteasomes. In yeast poly-ubiquitination seems to be achieved particularly by the E3 enzymes PEX2 and the E2 enzyme Ubc4p (or alternatively Ubc5p or Ubc1p), not PEX4. Mono-ubiquitination leads to delivery of the receptor to the cytosol for functioning in new rounds of PTS import. This modification involves PEX12 and PEX4. Before the receptor can mediate again in matrix protein import, its ubiquitin moiety has to be removed by a de-ubiquitinating enzyme. The enzyme responsible for this step has been identified in yeast as the PEX6-associated ubiquitin hydrolase Ubp15, while in mammals this function is accomplished by the cytosolic protease USP9X. For mammalian cells it has been proposed that the de-ubiquitination could also be achieved in a non-enzymatic manner, by a nucleophilic attack of glutathione. If a non-enzymatic mechanism also occurs in trypanosomatids, trypanothione might be involved.
The retrieval of the ubiquitinated PEX5 by PEX1 and PEX6 involves the hydrolysis of ATP. Interestingly, this is the only known step where energy is invested in the process, in addition to the activation of ubiquitin by an E1 enzyme. It implies that the import of proteins into the organelle's matrix is not achieved by direct energization of this univectorial process, but by coupling it to a cyclic vectorial process of the receptor that is energized in its export after having delivered its cargo into the matrix. An understanding of why this mechanism has evolved in this way can be obtained from the fact that this peroxisomal protein import is homologous, and seems to have been derived, from the ERAD process for retrieval of ubiquitinated, dysfunctional proteins from the ER lumen in which the energization also occurs by a cytosolic AAA+-ATPase (Schliebs et al. Reference Schliebs, Girzalsky and Erdmann2010).
The basic mechanism of matrix protein import seems conserved between peroxisomes of yeasts, mammals and other eukaryotes as well as glycosomes of trypanosomatids. For the major peroxins involved (PEX5, PEX7, PEX13, PEX4, PEX2, PEX10, PEX12, PEX1 and PEX6) homologues have been found in T. brucei (Fig. 2) and other trypanosomatids, although with low to very low similarity (35–15% sequence identity or less). Moreover, for each of these T. brucei peroxins the role in glycosome biogenesis has been confirmed (Moyersoen et al. Reference Moyersoen, Choe, Fan, Hol and Michels2004; Galland and Michels, Reference Galland and Michels2010). Ubiquitination of PEX5 and the involvement of the different peroxins in it have also been demonstrated (M.G.L. and P.M., unpublished data). Nonetheless, in several aspects considerable differences can be found between glycosome biogenesis in trypanosomatid parasites and peroxisome biogenesis in other eukaryotes. These differences, together with the low sequence conservation of the peroxins and the fact that glycosome biogenesis has been shown, by RNAi, to be a vital process and each of the peroxins to be essential, offers prospects for drug discovery (Moyersoen et al. Reference Moyersoen, Choe, Fan, Hol and Michels2004; Sampathkumar et al. Reference Sampathkumar, Roach, Michels and Hol2008; Galland and Michels, Reference Galland and Michels2010; Gualdrón-López et al. Reference Gualdrón-López, Michels, Quiñones, Cáceres, Avilán, Concepción, Jäger, Koch and Flohé2012c).
SOLUTE TRANSLOCATION THROUGH THE GLYCOSOMAL MEMBRANE
The compartmentalization of a considerable number of pathways, parts of pathways and even single enzymes not being part of a pathway within glycosomes necessitates the translocation of a large variety of solutes, substrates and products, through the boundary membrane of these organelles. These may be compounds of different sizes and charges, or neutral such as hexoses and glycerol. For example, many of the metabolic intermediates that have to be exchanged between glycosomal matrix and cytosol are phosphorylated, such as 3PGA, PEP, G3P, DHAP and others. Similarly, there will be a need for passage of inorganic ions, such as phosphate and pyrophosphate, Mg2+ and probably several others, cofactors and maybe also molecules that regulate the activity of enzymes and rates of processes inside the organelle. This exchange of solutes suggests a requirement for transporter molecules.
Only very few studies have been performed on the permeability of the glycosomal membrane in trypanosomatids and the translocation of solutes across it. These studies have been limited to T. brucei and performed only very recently. However, more information is available from research on peroxisomes from mammalian and plant cells and from yeast. The results thus obtained have guided the recent work on glycosomes.
Both the peroxisomal and glycosomal membrane form an apparent permeability barrier for solutes. Evidence for this notion is provided by the existence of mechanisms for the exchange of a reduced versus an oxidized solute such as malate/oxaloacetate and G3P/DHAP in membranes of both peroxisomes and glycosomes, respectively (Opperdoes and Borst, Reference Opperdoes and Borst1977; Van Roermund et al. Reference Van Roermund, Elgersma, Singh, Wanders and Tabak1995). Such an exchange is part of a system that shuttles electrons from NADH in the matrix of the organelle to the cytosol or mitochondria, to reoxidize the intraorganellar cofactor that itself cannot cross the membrane. Implicit in such a shuttle is the strict requirement for equimolecular exchange of solutes across the membrane, suggestive of the involvement of a specific transporter (Opperdoes and Borst, Reference Opperdoes and Borst1977; Bakker et al. Reference Bakker, Michels, Opperdoes and Westerhoff1997). Other arguments for the notion of the peroxisomal membrane as a permeability barrier are reports about the existence of transmembrane membrane potentials and pH gradients. The different arguments have previously been summarized (Wanders and Tager, Reference Wanders and Tager1998).
In the case of glycosomes, 3 additional findings, described in detail above, support the notion of the membrane as a permeability barrier. First, the possibility to reverse the GK reaction by accumulation of G3P and a low ATP/ADP ratio inside glycosomes (Opperdoes and Borst, Reference Opperdoes and Borst1977; Hammond et al. Reference Hammond, Aman and Wang1985; Bakker et al. Reference Bakker, Michels, Opperdoes and Westerhoff1997; Haanstra et al. Reference Haanstra, Van Tuijl, Kessler, Reijnders, Michels, Westerhoff, Parsons and Bakker2008). Second, the indication for the existence of different pools of glycolytic intermediates, in glycosomes and cytosol, which equilibrate only slowly, as obtained in the pulse-labelling experiment with radioactive glucose (Visser et al. Reference Visser, Opperdoes and Borst1981). Third, the prediction from computer simulation that T. brucei glycolysis cannot function if not separated from the cytosol, because of the peculiar kinetic properties of the enzymes, notably the kinases, and the experimental validation of this prediction (Bakker et al. Reference Bakker, Mensonides, Teusink, Van Hoek, Michels and Westerhoff2000; Furuya et al. Reference Furuya, Kessler, Jardim, Schnaufer, Crudder and Parsons2002; Haanstra et al. Reference Haanstra, Van Tuijl, Kessler, Reijnders, Michels, Westerhoff, Parsons and Bakker2008).
The impermeability of the membrane implies the need for transporters to translocate the specific solutes that should be able to cross the membrane. Indeed, 2 classes of solute transporters have been identified in peroxisomal membranes: ATP-binding cassette (ABC) transporters and mitochondrial carrier family (MCF) proteins (Theodoulou et al. Reference Theodoulou, Holdsworth and Baker2006; Visser et al. Reference Visser, Van Roermund, IJlst, Waterham and Wanders2007; Morita and Imanaka, Reference Morita and Imanaka2012). Mammalian, plant and yeast peroxisomes have each several isoforms of ABC transporters of the D-family in their peroxisomal membranes; these are in all cases half-size transporters containing only 1 integral membrane domain with 6 transmembrane helices and 1 nucleotide-binding domain exposed to the cytosol, in contrast to the more common ABC transporters which have 2 domains of each. Several of the peroxisomal ABC transporters, which mostly assemble as homodimers, have been shown to be involved in the uptake of long- and very long-chain acyl-CoAs and branched-chain acyl-CoAs. Whether some of the transporters also have other substrate specificities remains to be determined. Three half-size ABC transporters have also been found in trypanosomatids and, in the case of T. brucei, have been located in the glycosomal membrane (Yernaux et al. Reference Yernaux, Fransen, Brees, Lorenzen and Michels2006) (Fig. 3). One of them has been shown to be involved in the import of long-chain fatty acids such as oleoyl-CoA into the organelles (Igoillo-Esteve et al. Reference Igoillo-Esteve, Mazet, Deumer, Wallemacq and Michels2011). An MCF protein detected in the peroxisomal membrane of yeasts, plants and humans is an exchange carrier that seems to have a broad specificity for different compounds such as ATP, ADP, AMP, coenzyme A, FAD, FMN and NAD+ (Agrimi et al. Reference Agrimi, Russo, Scarcia and Palmieri2012). A full range of substrates and the relative affinities have only been assayed for the human MCF protein SLC25A17; the substrate specificity of the orthologous transporters of other organisms may differ. Indirect evidence for an adenine-nucleotide exchange transporter has been reported for T. cruzi glycosomes (Sanz-Rodríguez et al. Reference Sanz-Rodríguez, Concepción, Pekerar, Oldfield and Urbina2007). For T. brucei 24 MCF genes have been detected in its genome; so far 18 were located in the mitochondrion, and only 1 predominantly in the glycosomal membrane. But this latter one was only found in the glycosomes of the bloodstream-form stage, whereas it was detected mainly in the mitochondrial membrane in the procyclic form (Colasante et al. Reference Colasante, Alibu, Kirchberger, Tjaden, Clayton and Voncken2006, Reference Colasante, Peña Diaz, Clayton and Voncken2009).
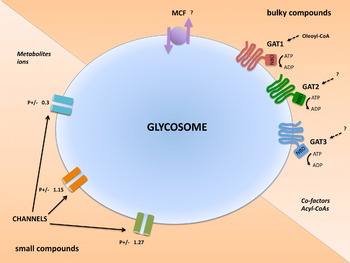
Fig. 3. Solute transporters and channels in the glycosomal membrane of Trypanosoma brucei. The membrane contains three half-size ABC transporters called GAT1-3, with a topology indicating their role as importers. GAT1 has been shown to transport oleoyl-CoA, the substrates of GAT2 and GAT3 remain to be identified. Probably, the membrane contains also MCF transporters, involved in transmembrane exchange of substrates. The ABC and MCF transporters are likely involved in transport of bulky (>500 Da) substrates such as cofactors [like ATP, ADP, NAD(P)] and acyl-CoAs. Additionally, glycosomal membrane preparations contain channel-forming activities suggesting the existence of at least three distinct channels with low substrate selectivity for non-bulky molecules such as glycolytic intermediates and ions. The ratio of selectivity for anions/cations (P + /–) is different for the distinct channels. Abbreviations: GAT, glycosomal ABC transporter, MCF, mitochondrial carrier family, NBD, nucleotide-binding domain.
The substrates of the peroxisomal and glycosomal solute transporters identified so far are all rather large molecules. On the contrary, all attempts thus far to find transporters for small molecules in any of these organelles have remained unsuccessful. However, based on transport studies with isolated mammalian peroxisomes it was already proposed in 1987 that the membrane contains general diffusion pores for small solutes (Van Veldhoven et al. Reference Van Veldhoven, Just and Mannaerts1987). Results of biochemical and electrophysiological experiments subsequently performed with plant peroxisomes also provided the conclusion that translocation of metabolites, such as carboxylates, across the membrane occurs via pores which represent relatively non-specific but highly efficient transport systems (reviewed by Reumann, Reference Reumann2000). More recently, studies with mammalian and yeast peroxisomes and solubilized peroxisomal membrane proteins reconstituted in artificial lipid membranes confirmed the existence of pores in the boundary membranes of the organelles from these organisms as well (Antonenkov et al. Reference Antonenkov, Sormunen and Hiltunen2004, Reference Antonenkov, Rokka, Sormunen, Benz and Hiltunen2005, Reference Antonenkov, Mindthoff, Grunau, Erdmann and Hiltunen2009; Antonenkov and Hiltunen, Reference Antonenkov and Hiltunen2012; Grunau et al. Reference Grunau, Mindthoff, Rottensteiner, Sormunen, Hiltunen, Erdmann and Antonenkov2009). These pores allow the permeation of small metabolites but restrict the exchange of more bulky compounds such as NAD(P)(H), ATP, ADP and AMP, CoA, acetyl/acyl derivatives, etc. These results, together with the identification and characterization of the transporters mentioned above, suggest that small solutes such as inorganic ions and most hydrophilic cellular metabolites can cross the membrane through general diffusion pores which, however, prevent the diffusion of bulky solutes such as cofactors and acyl-CoAs. The latter require transporter molecules.
A protein, Pxmp2, responsible for the formation of pores has recently been identified in murine peroxisomes (Rokka et al. Reference Rokka, Antonenkov, Soininen, Immonen, Pirilä, Bergmann, Sormunen, Weckström, Benz and Hiltunen2009). Studies with the reconstituted recombinant protein and studies with peroxisomes from wild-type and Pxmp2 −/− knockout mice showed that it forms a general diffusion pore. The channel formed by the pore has an estimated diameter of 1·4 nm, forming a size-selective filter with an exclusion limit of approximately 600 Da for hydrophilic solutes.
Recently, we have also found evidence for the existence of pores in the glycosomal membrane of trypanosomes (Gualdrón-López et al. Reference Gualdrón-López, Vapola, Miinalainen, Hiltunen, Michels and Antonenkov2012b) (Fig. 3). Solubilized membrane proteins from glycosomes purified from bloodstream-form T. brucei showed the capacity to form current-conducting pores when incorporated into artificial planar lipid membranes. Three main channel-forming activities were detected, each with a different conductance. No evidence for gating of the pores was obtained; the results predict that the channels form open, water-filled pores. One channel displayed a 3-fold higher selectivity for anions over cations, while the other two were slightly selective for cations. The anion-selective channel showed an intrinsic current rectification suggestive of a functional asymmetry of the pore in the membrane. The identity of the channel-forming proteins remains to be determined. In addition, the question of size-selection and solute specificities or preferences of the different pores, whether the pores are similarly expressed in different life-cycle stages of the parasite, and whether similar or other pores are present in glycosomal membranes of other trypanosomatid parasites also need to be determined. No homologues of the mammalian Pxmp2 could be found in any of the trypanosomatid sequence databases.
THE GLYCOSOMAL PARADOX
Several observations – the slowly equilibrating metabolite pools, reverse activity of GK, essentiality of compartmentalization of glycolysis – have indicated the existence of a permeability barrier between glycosome and cytosol. As described in the previous section, we have somewhat paradoxically found strong evidence for the existence of non-selective channels in the glycosomal membrane. These are similar to those detected in the membrane of peroxisomes from other organisms (Gualdrón-López et al. Reference Gualdrón-López, Vapola, Miinalainen, Hiltunen, Michels and Antonenkov2012b; Antonenkov and Hiltunen, Reference Antonenkov and Hiltunen2012). One possible explanation for this apparent paradox is that the slow equilibrium of metabolites across the membrane is due to the presence of channelling of these intermediates between active sites of enzymes catalysing consecutive reactions within the glycosomes (Fig. 4A). In this way the intermediate metabolites do not form part of a homogeneous pool within the entire organelle and consequently do not readily diffuse into the cytosol. Channelling may offer the advantage of decreasing transit times of intermediates and to enhance metabolic fluxes. A corollary is that it may exclude inhibitors from the active site of enzymes. A prerequisite of channelling is the need for organization of enzymes, either as stable multiprotein complexes or by transient protein-protein interactions between enzymes within a common metabolic pathway, forming what is called a metabolon (Srere, Reference Srere1987). Although there is strong evidence for channelling in several pathways (Vélot and Srere, Reference Vélot and Srere2000; Cleg and Jackson, Reference Clegg and Jackson1990), it remains in general difficult to demonstrate that channelling occurs and/or that enzymes are organized as functional multiprotein complexes. There is a general consensus that many protein-protein interactions are fragile and lost by dilution either during isolation or under in vitro conditions (Williamson and Sutcliffe, Reference Williamson and Sutcliffe2010). Despite this difficulty, there is increasing evidence that, indeed, enzyme-enzyme interactions exist in many metabolic pathways of different organisms (Beeckmans and Kanarek, Reference Beeckmans and Kanarek1987; Meyer et al. Reference Meyer, Gerwig, Hammer, Herzberg, Commichau, Völker and Stülke2011; Islam et al. Reference Islam, Nautiyal, Wynn, Mobley, Chuang and Hutson2010; Jørgensen et al. Reference Jørgensen, Rasmussen, Morant, Nielsen, Bjarnholt, Zagrobelny, Bak and Møller2005). Moreover, structural analysis of several enzyme complexes has shown molecular evidence for channelling such as a tunnel in tryptophan synthase (Hyde et al. Reference Hyde, Ahmed, Padlan, Miles and Davies1988) and other complexes (Miles et al. Reference Miles, Rhee and Davies1999). Another consequence of enzyme-enzyme interaction, different from channelling, is the alteration of kinetic properties of the enzymes involved (Ricard et al. Reference Ricard, Gontero, Avilán and Lebreton1998).

Fig. 4. Possible explanations for the ‘Glycosomal Paradox’. Three, not mutually exclusive explanations are proposed to explain how the apparent low permeability of the glycosomal membrane for metabolites is compatible with the existence of non-selective channels in the membrane. (A) Glycosomal enzymes form a multi-enzyme complex in which intermediates (I) in the conversion of substrate (S) to product (P) are channelled between active sites of consecutive enzymes of the pathway without major release into the lumen of the organelle and consequently through the channels into the cytosol. (B) Glycosomal enzymes have on average a higher pI than cytosolic proteins and are densely packed. The net positive charge of these proteins creates a Donnan equilibrium of charged molecules/ions across the glycosomal membrane, with the – mostly negatively charged – metabolites retained within the lumen. (C) Putative regulatory proteins (R) may control the opening and closing of the channels, or specific enzymes (E) at the beginning and end of the compartmentalized pathway may be associated with channels causing a preferential flux of the substrate from the cytosol directly into the active site of the enzyme, or a flux of the product from the active site into the cytosol.
Evidence for associations between enzymes of glycolysis, the most prominent pathway in the glycosome, has been reported for other cells. A complex of glycolytic enzymes formed by GAPDH, aldolase, PFK, PYK, lactate dehydrogenase and other proteins, was found in association with the membrane of red blood cells (Campanella et al. Reference Campanella, Chu and Low2005). The formation of this complex is regulated by oxygenation and phosphorylation. Furthermore, 6 glycolytic enzymes (aldolase, GAPDH, G3PDH, TPI, PGK and PGAM) co-localize at Z-disc and M-lines in Drosophila flight muscle (Sullivan et al. Reference Sullivan, MacIntyre, Fuda, Fiori, Barrilla and Ramizel2003). Glycolytic enzymes have also been found associated with other proteins such as tubulin (Vértessy et al. Reference Vértessy, Kovács and Ovádi1997) and calmodulin (Christova et al. Reference Christova, Orosz and Ovádi1996) among others. In some cases, interactions between glycolytic enzymes have been associated with modifications of their kinetic properties (Ovádi, Reference Ovádi1988).
Glycosomal proteins are found at a relatively high concentration inside glycosomes (approximately 150 mg/ml) of bloodstream-form T. brucei (Misset et al. Reference Misset, Bos and Opperdoes1986) and need an elevated concentration of salt (0·15–0·25 M NaCl) to be solubilized. Thus, one may speculate that a special organization exists within the organelle. Indeed, cross-linking experiments with T. brucei glycosomes have provided evidence that the enzymes are in close proximity, possibly with an average intermolecular distance of around 20 Å (Aman et al. Reference Aman, Kenyon and Wang1985).
The glycosomal enzymes have a tendency to associate and require high ionic strength during the purification process (Misset and Opperdoes, Reference Misset and Opperdoes1984). In addition, it has been shown that HXK exists as different aggregated forms at low ionic strength (Cáceres et al. Reference Cáceres, Portillo, Acosta, Rosales, Quiñones, Avilan, Salazar, Dubourdieu, Michels and Concepción2003). Another glycosomal enzyme, glucokinase, can be obtained as different molecular mass forms by gel filtration, dependent on the protein concentration at loading (Cáceres et al. Reference Cáceres, Quiñones, Gualdrón, Cordeiro, Avilán, Michels and Concepción2007). These behaviours would suggest a capacity of the glycolytic enzymes to make protein-protein interactions. However, evidence for channelling has not been found in this organelle. On the contrary, it has been argued that channelling is absent in the glycosome since a cross-linked complex and solubilized glycosome preparations had similar kinetic features (Aman and Wang, Reference Aman and Wang1986). In all, it is clear that more studies about the interactions of the glycosomal enzymes are necessary.
Another possibility to explain the low exchange of glycolytic intermediates across the glycosomal membrane, that is not mutually exclusive with the one discussed above, is the presence of a Donnan equilibrium over the membrane (Antonenkov and Hiltunen, Reference Antonenkov and Hiltunen2012) (Fig. 4B). The uneven distribution of charged macromolecules across 2 sides of a membrane that is only permeable for small compounds will lead to an asymmetrical distribution of permeant compounds with a charge opposite to that of the macromolecules. Matrix proteins of peroxisomes have usually a higher isoelectric point (pI) than those of the average cytosolic proteins and therefore are positively charged (Brett et al. Reference Brett, Donowitz and Rao2006). The pI's of many glycosomal proteins of trypanosomatids are even remarkably high (Misset et al. Reference Misset, Bos and Opperdoes1986; Hannaert and Michels, Reference Hannaert and Michels1994) which may lead to a tendency to restrict the diffusion of the negatively charged metabolites, such as the phosphorylated glycolytic intermediates from the matrix to the cytosol.
Another reason why diffusion of metabolites across the membrane is limited could be that the channels are associated with proteins that endow them with specificity and effectively control the passage of solutes. Such proteins may be regulatory factors or specific enzymes (Fig. 4C). For example, a specific association of HXK with a pore may make it specific for hexoses such as glucose. The glucose may then be immediately directed to the active site of HXK, resulting in a direct coupling of the translocation and ATP-dependent phosphorylation of the sugar. Similarly, an association of the glycosomal PGK with the same or another pore may make it specific for the efflux of its product 3-phosphoglycerate from glycosomes. In this respect, it may be relevant that, during purification, T. brucei HXK shows a strong tendency to remain associated with the glycosomal membrane (our unpublished results).
Although the equilibrium of glycolytic metabolites between the glycosomal matrix and cytosol, probably through channels in the glycosomal membrane, is slow compared to the glycolytic flux (60-fold slower in bloodstream-form T. brucei), it may be sufficient to sustain other metabolic processes in the cytosol that depend on such intermediates. For example, parallel routes of the PPP are present in glycosomes and cytosol (Michels et al. Reference Michels, Bringaud, Herman and Hannaert2006), but the substrate of this pathway, glucose 6-phosphate, seems synthesized exclusively by HXK – and in T. cruzi and Leishmania spp. also by a glucokinase (Cáceres et al. Reference Cáceres, Quiñones, Gualdrón, Cordeiro, Avilán, Michels and Concepción2007) – inside glycosomes and thus part of it has to exit the organelles. Another glycolytic intermediate of which small amounts should leave the glycosomes is fructose 6-phosphate that is phosphorylated by the cytosolic enzyme 6-phosphofructo-2-kinase to fructose 2,6-bisphosphate, the allosteric activator of PYK in trypanosomatids (Van Schaftingen et al. Reference Van Schaftingen, Opperdoes and Hers1985; Chevalier et al. Reference Chevalier, Bertrand, Rider, Opperdoes, Rigden and Michels2005).
GLYCOSOMES AND DRUG DISCOVERY
Glycosomal enzymes, notably those involved in glycolysis, are considered to be promising drug targets in T. brucei, but also in the other human pathogenic trypanosomatids, T. cruzi and Leishmania spp. (Verlinde et al. Reference Verlinde, Hannaert, Blonski, Willson, Périé, Fothergill-Gilmore, Opperdoes, Gelb, Hol and Michels2001; Opperdoes and Michels, Reference Opperdoes and Michels2001; Gualdrón-López et al. Reference Gualdrón-López, Michels, Quiñones, Cáceres, Avilán, Concepción, Jäger, Koch and Flohé2012c). Glycolysis is the only ATP-providing pathway in bloodstream-form T. brucei, and indeed almost all enzymes of the pathway have been genetically validated as drug targets (Albert et al. Reference Albert, Haanstra, Hannaert, Van Roy, Opperdoes, Bakker and Michels2005; Cáceres et al. Reference Cáceres, Michels and Hannaert2010). Glycolysis does probably not play a major role in the energy metabolism of Leishmania amastigotes living in the phagolysosomes of macrophages, where sugar levels are usually low, but formation of hexose 6-phosphate by gluconeogenesis as well as scavenging of exogenous hexoses and hexosamines and their phosphorylation appeared to be essential for survival in macrophages and establishment of normal infections (reviewed by McConville and Naderer, Reference McConville and Naderer2011 and Naderer and McConville, Reference McConville and Naderer2011). The phosphorylated sugar serves for essential processes such as the PPP, the synthesis of nucleic acids, myo-inositol and the reserve oligosaccharide mannogen and for the N-glycosylation of proteins to be expressed at the parasite's surface. Gluconeogenesis shares most of its enzymes with glycolysis and also occurs in glycosomes. For T. cruzi amastigotes living intracellularly in the cytosol of human host cells, the situation is probably very similar to that of Leishmania amastigotes, whereas the metabolism of T. cruzi blood trypomastigotes may be comparable to that of bloodstream-form T. brucei, as discussed elsewhere (Maugeri et al. Reference Maugeri, Cannata and Cazzulo2011; Gualdrón-López et al. Reference Gualdrón-López, Michels, Quiñones, Cáceres, Avilán, Concepción, Jäger, Koch and Flohé2012c).
Discovery of drugs targeted to these glycosomal enzymes seems realistic also when considering the long evolutionary distance between these protists and the human host, as well as the unique organization of these pathways inside the organelles, different from that in the host cells where they are in the cytosol (Gualdrón-López et al. Reference Gualdrón-López, Brennand, Hannaert, Quiñones, Cáceres, Bringaud, Concepción and Michels2012a). We have shown that this has endowed most of the trypanosomatid glycolytic/gluconeogenic enzymes with unique functional and structural properties that can be exploited for development of selective inhibitors (reviewed by Verlinde et al. Reference Verlinde, Hannaert, Blonski, Willson, Périé, Fothergill-Gilmore, Opperdoes, Gelb, Hol and Michels2001, Opperdoes and Michels, Reference Opperdoes and Michels2001 and Gualdrón-López et al. Reference Gualdrón-López, Michels, Quiñones, Cáceres, Avilán, Concepción, Jäger, Koch and Flohé2012c). Moreover, glycosomes harbour several other essential metabolic processes and possess many other enzymes that may be considered for drug discovery (Gualdrón-López et al. Reference Gualdrón-López, Michels, Quiñones, Cáceres, Avilán, Concepción, Jäger, Koch and Flohé2012c). Indeed, for several glycolytic and other glycosomal enzymes inhibitors have been developed that cause death of one or more of the in vitro-cultured trypanosomatid species, in many cases without affecting the corresponding host enzyme and growth of human cells, and for some compounds leading to a decrease of a parasitaemia in infected laboratory animals or even a cure (reviewed by Gualdrón-López et al. Reference Gualdrón-López, Michels, Quiñones, Cáceres, Avilán, Concepción, Jäger, Koch and Flohé2012c). A survey of such inhibitors is given in Table 1.
Table 1. Inhibitors of glycosomal glycolytic enzymes of trypanosomatids with trypanocidal effect

Note: The trypanocidal compound suramin inhibits almost all glycosomal glycolytic enzymes of T. brucei with IC50 values between 3 and 100 μM (Willson et al. Reference Willson, Callens, Kuntz, Périé and Opperdoes1993). However, it is doubtful that this drug kills the parasite by inhibition of glycolysis, because it is a large (MW 1297 Da), negatively charged molecule that probably does not easily crosses the glycosomal membrane. It has also an effect on many other enzymes, also mammalian glycolytic enzymes, and inhibits growth of cultured trypanosomes with an ED50 value as low as 50 nM.
Abbreviations: Tb, Trypanosoma brucei (bloodstream-form); Tc, Trypanosoma cruzi; Lm, Leishmania mexicana. IC50, concentration at which compound gives 50% activity inhibition in in vitro enzyme assays; ED50, concentration at which compound gives 50% growth inhibition of in vitro cultured parasites.
References: 1, Chambers et al. Reference Chambers, Kearns, Morris and Morris2008; 2, Dodson et al. Reference Dodson, Lyda, Chambers, Morris, Christensen and Morris2011; 3, Sharlow et al. Reference Sharlow, Lyda, Dodson, Mustata, Morris, Leimgruber, Lee, Kashiwada, Close, Lazo and Morris2010; 4, Hudock et al. Reference Hudock, Sanz-Rodríguez, Song, Chan, Zhang, Odeh, Kosztowski, Leon-Rossell, Concepción, Yardley, Croft, Urbina and Oldfield2006; 5, Sanz-Rodríguez et al. Reference Sanz-Rodríguez, Concepción, Pekerar, Oldfield and Urbina2007; 6, Nowicki et al. Reference Nowicki, Tulloch, Worrall, McNae, Hannaert, Michels, Fothergill-Gilmore, Walkinshaw and Turner2008; 7, Dax et al. Reference Dax, Duffieux, Chabot, Coincon, Sygusch, Michels and Blonski2006; 8, Azéma et al. Reference Azéma, Lherbet, Baudoin and Blonski2006; 9, Aronov et al. Reference Aronov, Suresh, Buckner, Van Voorhis, Verlinde, Opperdoes, Hol and Gelb1999; 10, Bressi et al. Reference Bressi, Verlinde, Aronov, Shaw, Shin, Nguyen, Suresh, Buckner, Van Voorhis, Kuntz, Hol and Gelb2001; 11, Bressi et al. Reference Bressi, Choe, Hough, Buckner, Van Voorhis, Verlinde, Hol and Gelb2000.
As argued above, correct compartmentalization of enzymes inside glycosomes and the biogenesis of the organelles are also essential for the parasites and the peroxins responsible for the formation of the organelles are poorly conserved compared to their homologues that mediate peroxisome biogenesis in human cells (Moyersoen et al. Reference Moyersoen, Choe, Fan, Hol and Michels2004; Galland and Michels, Reference Galland and Michels2010). This suggests ample opportunities for the exploitation of peroxins for anti-trypanosomatid chemotherapy. Contrary to glycolytic enzymes, only limited information is as yet available about the 3-dimensional structures of peroxins. Nonetheless, crystal structures of the human and T. brucei PTS1-protein binding C-terminal domain of PEX5 have been determined and a comparison suggests various possibilities for compounds that may selectively block PTS1-protein import into glycosomes (Gatto et al. Reference Gatto, Geisbrecht, Gould and Berg2000; Stanley and Wilmanns, Reference Stanley and Wilmanns2006; Sampathkumar et al. Reference Sampathkumar, Roach, Michels and Hol2008). The development of compounds interfering with matrix protein import into glycosomes is currently actively pursued by detailed structural studies of T. brucei peroxins involved in this process, and analysis of the interactions made between different peroxins during the successive stages of the PTS-protein translocation, as well as by screening of chemical compound libraries with recombinant peroxins. Promising preliminary results have been obtained.
The solute transporters and channels in the glycosomal membrane may also offer perspectives as drug targets. Although still very little is known about them – only 1 class of transporters has recently been recognized and partially characterized and the proteins responsible for channel formation remain to be identified – they may be considered as potential drug targets. Correct localization of glycosomal metabolism, or at least part of it, is essential for the parasites and therefore the translocation of the substrates and products across the membrane should be essential as well. Moreover, the glycosomal ABC transporters show very little sequence similarity with their counterparts in the peroxisomes or with other ABC transporters in human cells and probably the same will be true for the channel-forming proteins. No homologue of the currently only known mammalian peroxisomal channel protein can be found in the trypanosomatid genome databases.
An advantage of targeting transporters and channels in the glycosomal membrane is that they, or at least the solute binding part, may be readily available for inhibitors at the cytosolic face of the membrane. The same holds true for peroxins as drug targets. However, inhibitors of glycosomal enzymes have to enter the organelle. One could consider several possibilities by which these latter compounds cross the membrane: (1) using existing transporters; (2) by passive diffusion through the lipid bilayer; (3) via the channels (Fig. 5). The first option is not very likely. The number of different transporter molecules seems very limited. Although these transporters may have relatively broad substrate specificities, the structural diversity of inhibitors that may be transported is possibly very limited. The second option would possibly only be feasible for non-polar compounds; such compounds, like those obeying the ‘Lipinski rule of five’ for good solubility and permeability properties as found for many drugs (Lipinski et al. Reference Lipinski, Lombardo, Dominy and Feeney2001), may enter via this route. However, such properties may be difficult to combine with a high affinity for the target enzymes to be inhibited, most of them catalysing reactions with negatively charged substrates and therefore having several positively charged residues in their active sites. Moreover, recent analyses suggest that even most known hydrophobic drugs do not reach their intracellular targets by passively permeating through the lipid-bilayer membranes of cells, but are internalized by transporters (Dobson and Kell, Reference Dobson and Kell2008; Dobson et al. Reference Dobson, Lanthaler, Oliver and Kell2009; Lanthaler et al. Reference Lanthaler, Bilsland, Dobson, Moss, Pir, Kell and Oliver2011; Kell et al. Reference Kell, Dobson and Oliver2011). Also the recent finding that most known trypanocidal compounds enter the parasites via transporters and not passively is noteworthy in this respect (Schuman-Burkard et al. Reference Schumann-Burkard, Jutzi and Roditi2011; Alsford et al. Reference Alsford, Eckert, Baker, Glover, Sanchez-Flores, Leung, Turner, Field, Berriman and Horn2012). One has therefore also to consider the third option, i.e. that many inhibitors of glycosomal enzymes may have to enter the organelles through the channels that may impose little selectivity on the structure and charge of the compounds, only on their size. Assuming that the pores in the glycosomal membrane form channels with similar diameters as those in peroxisomal membranes (Antonenkov and Hiltunen, Reference Antonenkov and Hiltunen2012), it is expected that the molecular mass of the inhibitors should be limited to 400–500 Da. We assume that most, if not all currently available inhibitors of purified glycosomal enzymes with trypanocidal activity and which have been shown to most likely kill the cells indeed via these targets in situ (reviewed by Gualdrón-López et al. Reference Gualdrón-López, Michels, Quiñones, Cáceres, Avilán, Concepción, Jäger, Koch and Flohé2012c), will cross the glycosomal membrane via these channels. If indeed the channels play an important role in the targeting of glycosomal enzymes, the pore size sets a limit on the size of inhibitors to be developed in the future.

Fig. 5. Diagrams depicting how drugs for human sleeping sickness may reach their targets inside the glycosomes. Upper panel: drugs for stage 1 of the disease should preferably be orally administered, thus to be able to cross the intestinal epithelium. Lower panel: drugs for stage 2, when trypanosomes are present also in the interstitial fluid of the brain, should also be capable of crossing the blood-brain barrier (BBB). Green dots represent the drugs. Indicated are the different means by which the drugs may cross successive membranes. (1) By using transporter systems in the intestinal epithelium, the BBB and the plasma membrane of the trypanosome; (2) by passive diffusion through each of the successive membranes; (3) via the tight junctions between cells of the BBB; (4) in association with blood proteins (red dots) via receptor-mediated endocytosis through the flagellar pocket and the endosomal system of the trypanosome; (5) by fluid-phase endocytosis through the flagellar pocket; (6) by passive diffusion of pro-drugs (a) of which the charge is unmasked in the cytosol leading to activation of the drug (b); (7) by translocation of neutral (a) or charged (b) drugs via ABC or MCF transporters in the glycosomal membrane; (8) by translocation of neutral (a) or charged (b) drugs via one of the channels in the glycosomal membrane.
It is unlikely that the putative mechanism(s) that are responsible for the low equilibrium of the glycolytic and possibly other metabolic intermediates across the membrane, despite the existence of non-selective, water-filled pores, may also limit the uptake of drugs, i.e. a Donnan potential or control of the opening of the pore by regulatory proteins or association with metabolic enzymes. The Donnan potential will only have an effect on the entry of negatively charged inhibitors. In bloodstream-form T. brucei, the transmembrane equilibrium of metabolites is slow compared to the glycolytic flux, but very fast compared to the division time of a trypanosome. Furthermore, it is also conceivable that inhibitors can enter through the transient pore involved in the import of matrix proteins, in free form or bound to the newly synthesized target proteins that are imported. In this case there would be no limit on the size of the drug, but the occasion for entry into glycosomes is limited in time – during the occasional and short periods the pores are opened – and restricted to the fraction of glycosomes that are competent for matrix protein import.
The high intraglycosomal protein concentration, with the probability of the existence of a multi-enzyme complex and the possibility of metabolite channelling between the active sites of the enzymes in the complex may also have consequences for targeting drugs to these enzymes. Could the active sites inside such a complex easily be reached by the drugs? Does channelling of intermediates directly from the active site of one enzyme to the active site of the next one along a ‘highway’ not lead to shorter transit times and/or high concentrations of the intermediates with which drugs have to compete? These questions may be relevant for the choice of the enzyme to be targeted, and also for the structural and physicochemical properties of the inhibitor to be used as a drug, the affinity for its target and its mode of inhibition.
CONCLUSIONS AND PERSPECTIVES
In this review, we have summarized the results of our recent work that shown that, in many respects, solutes and proteins cross the membrane of glycosomes in trypanosomes (and the membrane of peroxisomes in other eukaryotes) by mechanisms quite different from those operating in other membranes such as mitochondria, chloroplasts and plasma membranes. In the case of protein import, similarity (and an evolutionary relationship) exists only with the system of protein retrieval from the lumen of the ER to the cytosol, generally present in eukaryotes. For the translocation of bulky solutes, these organelles employ half-sized ABC transporters (and also MCF transporters in peroxisomes, but this remains to be proven for glycosomes) belonging to a subfamily from a very large class of carrier molecules which are used by all organisms – pro- and eukaryotes – for a large variety of substrates. Smaller solutes seem to pass through the glycosomal/peroxisomal membranes by several distinct water-filled channels with no or only little selectivity. It is therefore inferred that the apparent low permeability for metabolites should be attributed to properties not directly related to the membrane, such as metabolite channelling within a multi-enzyme complex, the maintenance of a Donnan distribution across the membrane and/or proteins that regulate access of solutes to the channels.
Our knowledge of the mechanism of solute transport through the glycosomal membrane, and the identity of molecules responsible for it, is still very limited. Further studies are required to (i) identify all solute transporters; (ii) to determine the substrate specificities of the transporters; (iii) to characterize the channels, i.e. the identification of the proteins involved in their formation, the determination of their structures and their selectivity for solutes; (iv) to find one or different complementary explanations for the glycosomal paradox of the existence of channels and a slow transmembrane equilibrium of metabolites.
The proteins involved in translocation of molecules across the glycosomal membrane may be considered as potential drug targets. This holds true for both the peroxins involved in the import of glycosomal matrix proteins as well as for at least some of the transporters and channels responsible for translocation of cofactors and metabolites. Glycosomal biogenesis is vital for the parasites and drugs that disrupt or prevent the interaction of peroxins in the cascade of reactions resulting in the delivery of PTS proteins to the matrix will therefore kill the trypanosomes. Of course, peroxisomes play also important roles in human cells, but in many respects these are different from those of glycosomes in the parasites. Although currently only very little information is available about three-dimensional structures of peroxins, the low – in some cases even very low degree of similarity – in primary structures between trypanosomes and human peroxins, and the differences observed in interfaces of interacting peroxins offer promise that compounds will be discovered that selectively interfere with glycosome formation (Galland and Michels, Reference Galland and Michels2010). Similarly, glycosomal metabolism is essential and thus the correct translocation of substrates and products of enzymes or pathways is vital. There exists only very low sequence similarity of known glycosomal transporter molecules with their human counterparts and no homologue could be discovered in the trypanosomatid databases for the only known human peroxisomal channel-forming protein. Therefore, it seems very likely that compounds could be obtained that selectively inhibit the parasite transporters or block their channels with no effect on the function of human peroxisomal metabolism.
How will trypanocidal drugs that are meant to act by inhibiting glycosomal enzymes reach their target? As we argued above, passage through the glycosomal membrane channels seems a very attractive option, probably only setting an upper limit on the size of the molecule of about 400–500 Da. However, this criterion is not unique; it is also one of the criteria in the ‘Lipinski rule of five’. Compounds destined for the glycosome should, however, first cross also other membranes like the parasite's plasma membrane (Fig. 5). Recent studies have shown that, in general, most drugs enter cells via transporters or receptors (Lanthaler et al. Reference Lanthaler, Bilsland, Dobson, Moss, Pir, Kell and Oliver2011; Kell et al. Reference Kell, Dobson and Oliver2011). This is also the case for many known trypanosomatid drugs: melarsoprol and diamidines via the aminopurine P2 transporter, eflornithine via the amino-acid permease TbAAT6, antimonials via aquaglyceroporins and suramin by receptor-mediated endocytosis with the invariant surface glycoprotein ISG75 (Schuman-Burkard et al. Reference Schumann-Burkard, Jutzi and Roditi2011; Alsford et al. Reference Alsford, Eckert, Baker, Glover, Sanchez-Flores, Leung, Turner, Field, Berriman and Horn2012). Only nifurtimox probably does diffuse through the membrane. Furthermore, new drugs to be developed for stage 1 of sleeping sickness should preferably given orally, thus must be able to be taken up via the intestine (Fig. 5A), whereas drugs for stage 2 of the disease have to cross also the blood brain barrier (Fig. 5B). In that case design of Lipinski compliant compounds might be the best option, because that would allow them to cross this latter barrier as well as successively all other membranes (Wager et al. Reference Wager, Villalobos, Verhoest, Hou and Shaffer2011; Ghose et al. Reference Ghose, Herbertz, Hudkins, Dorsey and Mallamo2012). However, recent measurements indicate that, contrary to the widespread notion, such passive permeation is very slow (Kell et al. Reference Kell, Dobson and Oliver2011). An alternative scenario, notably to be considered for charged compounds, is to let them enter the parasite by endocytosis, bound to macromolecules or in bulk liquid phase, or by passive diffusion as a neutral pro-drug, in which the charged group is masked, but that will be de-protected by appropriate enzymes (esterases, phosphodiesterases or reductases, dependent on how the protecting group is linked to the drug) in the trypanosome's cytosol, and then enter the glycosomes via channels. The pro-drug scenario may be suitable for killing bloodstream T. brucei and T. cruzi trypomastigotes with glycosomal enzyme inhibitors, but not for the intracellular amastigotes of T. cruzi and Leishmania spp., because the masking group of the pro-drugs may already be removed by the activity of hydrolases in the cytosol of the host cell, thus exposing the charged group and consequently rendering the drug incapable of crossing the plasma membrane of the parasite. Indeed, it has been shown that pro-drug forms of an effective inhibitor of the glycosomal enzyme aldolase kills cultured bloodstream-form T. brucei efficiently, but have no effect on intracellular amastigotes of Leishmania infantilis and T. cruzi (Azéma et al. Reference Azéma, Lherbet, Baudoin and Blonski2006).
With all these considerations about crossing membranes, one may wonder whether enzymes inside glycosomes are indeed such suitable drug targets against trypanosomes. We do indeed consider that they are. First because, as discussed above, glycolytic enzymes are essential for the parasites and have structural and functional features that allow the discovery of parasite enzyme selective inhibitors, as has been amply demonstrated (reviewed by Verlinde et al. Reference Verlinde, Hannaert, Blonski, Willson, Périé, Fothergill-Gilmore, Opperdoes, Gelb, Hol and Michels2001, Opperdoes and Michels, Reference Opperdoes and Michels2001 and Gualdrón-López et al. Reference Gualdrón-López, Michels, Quiñones, Cáceres, Avilán, Concepción, Jäger, Koch and Flohé2012c). Moreover, some of the inhibitors kill in vitro cultured bloodstream-form T. brucei with no or little effect on growth of human cells (Gualdrón-López et al. Reference Gualdrón-López, Michels, Quiñones, Cáceres, Avilán, Concepción, Jäger, Koch and Flohé2012c). Furthermore, reduction of the glycolytic flux by 50%, what can be achieved by partial inhibition of enzymes, is already sufficient to kill the parasites (Albert et al. Reference Albert, Haanstra, Hannaert, Van Roy, Opperdoes, Bakker and Michels2005; Haanstra et al. Reference Haanstra, Kerkhoven, Van Tuijl, Blits, Wurst, Van Nuland, Albert, Michels, Bouwman, Clayton, Westerhoff and Bakker2011). And in several cases it has been proven, or shown to be very likely, that inhibitors that killed the parasites indeed had been able to reach enzymes inside glycosomes, such as aldolase, GAPDH and HXK (Aronov et al. Reference Aronov, Suresh, Buckner, Van Voorhis, Verlinde, Opperdoes, Hol and Gelb1999; Dax et al. Reference Dax, Duffieux, Chabot, Coincon, Sygusch, Michels and Blonski2006; Azéma et al. Reference Azéma, Lherbet, Baudoin and Blonski2006; Chambers et al. Reference Chambers, Kearns, Morris and Morris2008; Dodson et al. Reference Dodson, Lyda, Chambers, Morris, Christensen and Morris2011; Sharlow et al. Reference Sharlow, Lyda, Dodson, Mustata, Morris, Leimgruber, Lee, Kashiwada, Close, Lazo and Morris2010) (see also Table 1). After treatment of trypanosomes with the cymelarsan (melarsamine hydrochloride), this compound was found covalently bound to glycosomal G3PDH and to inhibit the enzyme (Denise et al. Reference Denise, Giroud, Barrett and Baltz1999), although it is doubtful if this enzyme is the most important target of this known trypanocidal arsenical drug (Foucher et al. Reference Foucher, McIntosh, Douce, Wastling, Tait and Turner2006). We hypothesize that most of these inhibitors are able to enter the glycosomes through the channels, because of their relatively low molecular weights (<500 Da). Indeed, for the fluorescent HXK inhibitor quercetin (MW 338 Da) the glycosomal localization was confirmed (Dodson et al. Reference Dodson, Lyda, Chambers, Morris, Christensen and Morris2011). The larger GAPDH inhibitors are adenosine analogues (MW ∼540 Da) and may be transported by a still to be identified transporter with broad specificity for cofactors.
In conclusion, we consider that recent investigations on protein and solute translocation across the glycosomal membrane have revealed new opportunities for drug discovery, but have also provided new information that may allow us refine the criteria for the size and chemical nature of the drugs to be developed. Moreover, if indeed future research will confirm the presence of an intraglycosomal multi-enzyme complex in which metabolites are channelled, as an explanation of the glycosomal paradox, it will be relevant to consider also this aspect in the development of inhibitors that will be working optimally in situ against such enzymes and therefore will offer good prospects to be used as leads for anti-trypanosomatid drugs.
ACKNOWLEDGEMENTS
We thank Drs Jurgen Haanstra (University of Groningen), Malcolm Walkinshaw, Linda Fothergill-Gilmore and Hugh Morgan (University of Edinburgh), Wim Hol (University of Washington, Seattle) and Michael Barrett and Fiona Achcar (University of Glasgow) for helpful discussions and/or providing critical comments on an earlier version of the manuscript.
FINANCIAL SUPPORT
M.G.L. and A.B. gratefully acknowledge their Ph. D. scholarships from the ‘Fonds pour la formation à la Recherche dans l'Industrie et dans l'Agriculture’ (FRIA) and the ‘Patrimoine de Faculté Médicine, Université catholique de Louvain’. The work of L.A. was financially supported by the ‘Fondo Nacional de Ciencia, Tecnología e Innovación’ (FONACIT) in project MC-2007000960 and that of P.A.M.M. by the ‘Fonds de la Recherche Scientifique’ (F.R.S.-FNRS) and the Belgian Interuniversity Attraction Poles-Federal Office for Scientific, Technical and Cultural Affairs.






