INTRODUCTION
A proteome is the network of proteins that comprise a biological system. Proteomics, a concept first enunciated just 15 years ago (Wilkins et al. Reference Wilkins, Pasquali, Appel, Ou, Golaz, Sanchez, Yan, Gooley, Hughes, Humphery-Smith, Williams and Hochstrasser1996), developed from genomics and thus has the global aim of reporting on all proteins in a system such as a cell or organism. This is an ambitious goal as even relatively simple organisms express thousands of proteins. The challenge is amplified when, as is often the case, there is a need to dissect a particular phenotype by monitoring the relative abundance of proteins or by defining post-translational protein modifications. The time and technology that must be invested to approach a global proteomic analysis is beyond the resource of many research groups, but many of the same proteomic technologies can be applied in a targeted way to a specific sub-proteome. This review aims to introduce the key technologies that enable proteomics and to illustrate how these approaches can be focused on some of the biological questions that typically exercise parasitologists.
Proteomic analysis has the potential to reveal the mechanisms through which organisms develop and respond to environmental challenges. Since pathogens interact with their hosts via proteins and their products, proteomic analysis has clear potential to elucidate the mechanisms by which parasites cause disease, as well as the responses that are elicited in the host. Most drugs target proteins and proteomic analyses can thus contribute to the characterization of new drug targets, as well as the elucidation of the targets of, and resistance mechanisms to, existing drugs.
A variety of sophisticated procedures has been developed to monitor, identify, quantify and characterize specific proteins of interest. Most of these approaches are predicated on some prior knowledge of the protein in question (function, antigenic properties for example) and cannot report on proteins that are not targeted for analysis, or for which specific tools (such as enzyme assays and antibodies) are not available. Furthermore, classical approaches to protein characterization are reductionist, and have limited application where the goal is to characterize a protein network or to draw inference about the biological system.
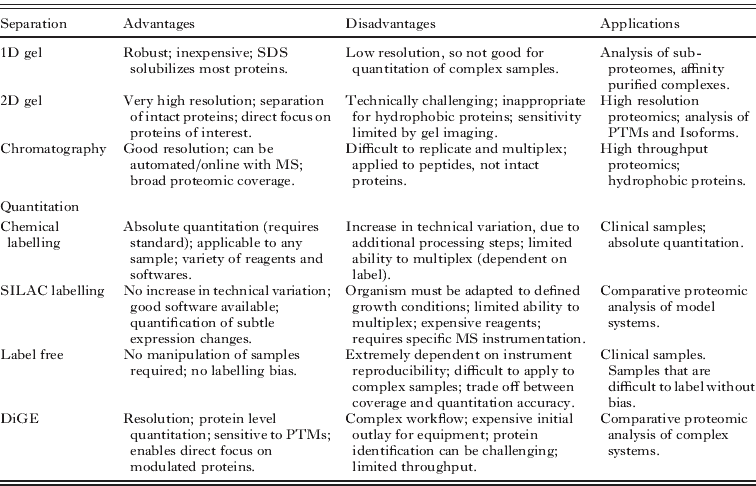
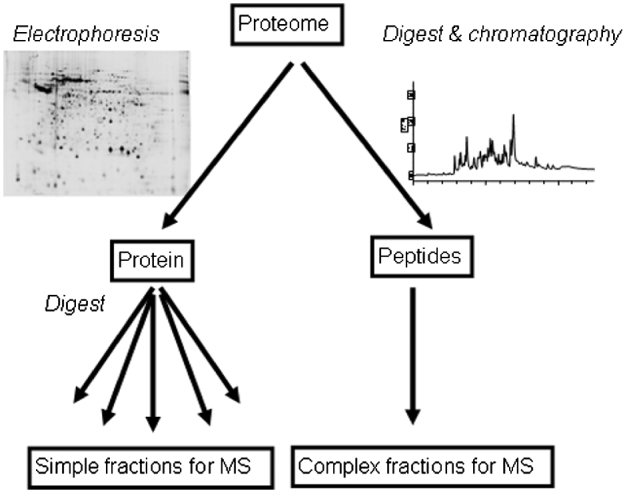
Conversely, proteomics holds forth the promise of the simultaneous characterization of all of the proteins in a system. The processes of transcription, translation and maturation by which genes give rise to proteins are highly dynamic, so proteomes are complex and labile. Thus true global proteomics is an ambitious goal even for the simplest organisms, but a proteomics approach can realistically report on a subset of typically more abundant and more soluble proteins. The data obtained can be sufficient to enable systems modelling and to direct subsequent more targeted studies. Proteomics can also be focused on relatively restricted sub-proteomes, such as affinity-purified protein complexes or enriched organellar fractions. Such targeted approaches can be more achievable but require appropriate biochemical tools and strategies to generate a sub-proteome that comprises relevant proteins. The application of proteomic technologies to subcellular fractions can reveal new components without preconception, making this a powerful approach in cell biology. Post-translational modifications can potentially be resolved or characterized and quantitative approaches can elucidate stoichiometry and reveal regulatory changes. Proteomic analyses require complex and expensive mass spectrometry as well as some proteomic expertise and, in many cases, acutely targeted approaches such as Western blotting and activity assays are sufficient to monitor the expression of proteins of interest. However, proteomics can also enable discovery of new proteins of interest (Fig. 1).

Fig. 1. Relationship between proteomic sample complexity and proteomic separation method. Proteomes must be resolved into their components so that individual protein species can be highlighted and identified. Both gel electrophoresis (gel) and liquid chromatography (LC) can be employed, in single (1D) or multiple (2D) dimensions. The combination of protein and peptide separation approaches that are employed depends upon the complexity of the proteome under analysis, and upon the method selected for relative quantitation.
PROTEIN IDENTIFICATION BASICS
Proteomics involves protein or peptide separation, with the aim of protein identification and often the inference of characteristics such as abundance, localisation or modification. Ideally, the proteome of interest should be resolved into components that are amenable to characterization by mass spectrometry (Table 1). Protein identification by mass spectrometry may involve simple measurement of the mass of tryptic digest peptides, giving information that reflects amino acid composition and can enable protein identification by peptide mass fingerprinting (Pappin et al. Reference Pappin, Hojrup and Bleasby1993, Reference Pappin1997). This approach is generally useful for proteomics only when applied to a relatively pure species that is derived from an organism for which genome sequence data are available, because it is required that multiple peptide masses, from a list of limited length, match with statistical significance to a specific parent protein. Peptide mass fingerprinting will likely fail if the list of peptide masses that derive from the mass spectrometer is too long or contains masses that match to multiple gene products. Furthermore, because amino acid substitution will alter the observed peptide mass, no identification will be obtained if there is no very similar protein represented in available databases. This limitation generally hampers protein identification between species or even strains. Genome sequence data are unavailable or incomplete for many parasites and so peptide mass fingerprinting approaches have limited utility in parasite proteomics.
Table 1. Relative merits of typical proteomic separation and quantitation strategies
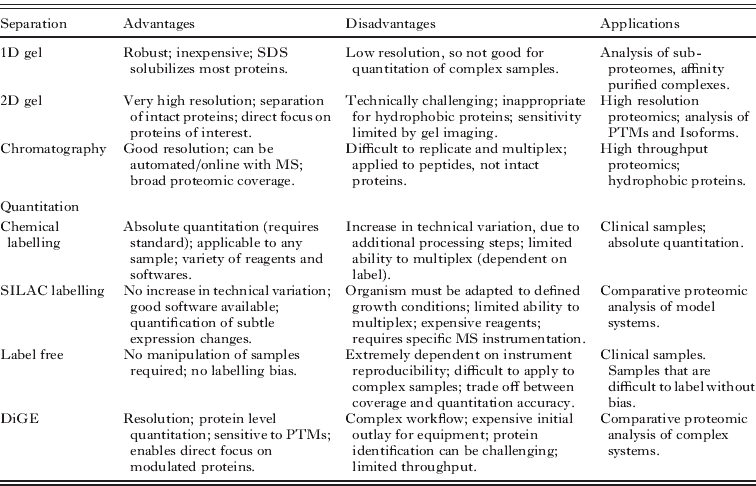
PTM – Post-translational modification; SILAC – Stable Incorporation of Amino acids in Culture; DiGE – Difference Gel Electrophoresis.
More sophisticated tandem mass spectrometry (MS/MS) can circumvent many of these issues by providing information on both amino acid composition and sequence for some peptides. Combined amino acid composition and sequence information can permit confident protein identification based on characterization of a limited number of peptides and database matching can be performed despite some divergence between the organism of interest and the most homologous genome sequence database available (Hernandez et al. Reference Hernandez, Muller and Appel2006). However, tandem mass spectrometers generally select ions individually for fragmentation analysis and peptides from a mixed population are generally selected on the basis of abundance. Thus it is more efficient to separate peptides prior to their introduction to the mass spectrometer, and this is particularly so when the peptide mixture under study is derived from the digest of mixed proteins. It is important to present any mass spectrometer with relatively simple mixtures, to allow an output that represents the masses of as many constituents as possible, in a format that can be deconvoluted and matched to genome datasets.
Since a proteome is, by definition, a complex mixture of proteins, it is critical to adopt a high resolution separation workflow so that the proteome is fractionated prior to mass spectrometry analysis. Furthermore, proteomes typically comprise a broad dynamic range, with some superabundant components and many that are of much lower abundance (Corthals et al. Reference Corthals, Wasinger, Hochstrasser and Sanchez2000). Fractionation can permit the characterization of relatively low abundance components, by separating them from abundant components.
Protein separation
The separation of complex protein mixtures is typically performed on intact proteins by electrophoresis or on peptides by chromatography or within the mass spectrometer. For even relatively simple proteomes, the application of each of these separations in series is often required to maximise the proportion of the proteome that is ultimately characterized. One-dimensional gel electrophoresis (1-DE), almost always sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) (Laemmli, Reference Laemmli1970), is a relatively low resolution technique but is a valuable separation approach in proteomics because it is robust and because SDS is a strong ionic detergent that solubilises relatively hydrophobic proteins. 2-dimensional gel electrophoresis (2-DE) brings much higher resolution. Classically, the 2 electrophoretic dimensions employed are isoelectric focusing and SDS-PAGE (O'Farrell, Reference O'Farrell1975). In large format gel systems, classical 2-DE can resolve several thousand protein species, a resolving power that is unsurpassed in proteomic workflows. However, 2-DE is a separation approach that is technically challenging and can therefore be difficult to replicate and time consuming to perform. Furthermore, the requirement to limit conductivity in isoelectric focusing means that ionic detergents such as SDS cannot be employed. This constraint means that classical 2-DE separation does not resolve the hydrophobic integral membrane proteins that play a critical role in biology (Pedersen et al. Reference Pedersen, Harry, Sebastian, Baker, Traini, McCarthy, Manoharan, Wilkins, Gooley, Righetti, Packer, Williams and Herbert2003). Alternative 2-DE separations have been developed with the aim of enhancing the ability to resolve hydrophobic proteins (Schagger and van Jagow, Reference Schagger and von Jagow1991; Appel et al. Reference Appel, Hochstrasser, Funk, Vargas, Pellegrini, Muller and Scherrer1991; Hartinger et al. Reference Hartinger, Stenius, Hogemann and Jahn1996; Bridges et al. Reference Bridges, Pitt, Hanrahan, Brennan, Voorheis, Herzyk, de Koning and Burchmore2008). Although effective, such approaches are generally less highly resolving than classical 2-DE. Despite these limitations, which have led many researchers to discount the use of 2-DE in proteomics, the ability to fix and stain gels to visualise separated proteins brings several advantages, including the ability to infer the relative abundance of protein components, to resolve protein species that bear specific post-translational modifications and to focus subsequent protein identification efforts directly on proteins of interest, by excising regions of the gel.
Chromatography represents the most popular alternative to gel electrophoresis for proteomic separation (Wolters et al. Reference Wolters, Washburn and Yates2001; Hall et al. Reference Hall, Karras, Raine, Carlton, Kooij, Berriman, Florens, Janssen, Pain, Christophides, James, Rutherford, Harris, Harris, Churcher, Quail, Ormond, Doggett, Trueman, Mendoza, Bidwell, Rajandream, Carucci, Yates, Kafatos, Janse, Barrell, Turner, Waters and Sinden2005; Panchaud et al. Reference Panchaud, Scherl, Shaffer, von Haller, Kulasekara, Miller and Goodlett2009). Chromatographic separation of intact proteins is generally not used in proteomic workflows, in part because of the broad heterogeneity of intact proteins. Instead, chromatography is usually applied after digest (Fig. 2). Like gel electrophoresis, the resolution of chromatography can be greatly enhanced by the establishment of multiple, orthogonal separations. For example, ion exchange chromatography can precede reversed phase chromatography to achieve 2 dimensional chromatographic separation (Wolters et al. Reference Wolters, Washburn and Yates2001). The increase in complexity that is introduced when proteins are converted to peptides, together with the necessity to collect fractions between the first and second dimensions of chromatography, means that the proteomic resolution of 2-D electrophoresis remains superior to 2-D chromatography (Bridges et al. Reference Bridges, Pitt, Hanrahan, Brennan, Voorheis, Herzyk, de Koning and Burchmore2008). However, the significant resolving power of capillary flow HPLC, together with the capacity for automation, means that chromatography is overtaking classical 2-DE as the workhorse separation for proteomics (Xie et al. Reference Xie, Liu, Qian, Petyuk and Smith2011).
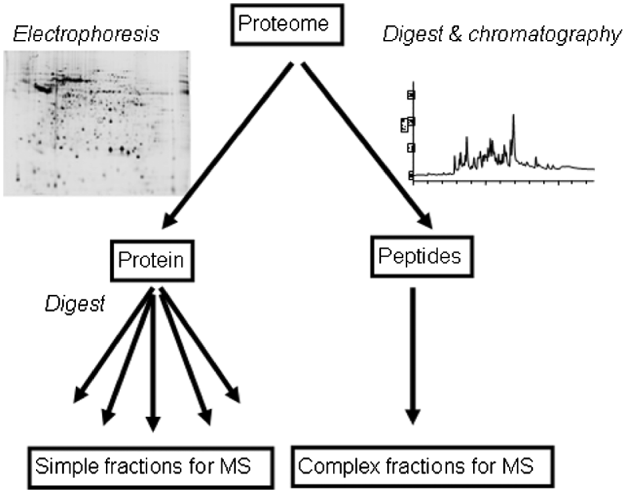
Fig. 2. Gel-based versus gel-free proteomic workflows. Gel-based proteomic separations are applied to the intact proteins, whilst gel-free separation is applied to peptides.
The separation workflow that is most commonly employed involves a combination of electrophoresis and chromatography, and it exploits the complementary advantages of each approach. SDS-PAGE is typically applied to separate intact proteins in a mixture. Fractions are then excised from the gel lane and the proteins digested to produce peptides that are separated by reversed phase chromatography, representing the second dimension of separation. The C18 resins that are generally used as the stationary phase separate small peptides of the type generally produced by tryptic digest with high resolution. Providing an appropriate wash step has been included, the eluent from reversed phase chromatography has low ionic content and is suitable for direct infusion into a mass spectrometer with an electrospray ionisation source. Current tandem mass spectrometers can operate sufficiently quickly to achieve a third dimension of separation, by analysing multiple species sequentially but in a time significantly lower than the typical HPLC peak widths. This workflow, often called Gel-C-MS, can be semi-automated and represents an efficient use of instrument time.
Quantitative proteomics
One of the most powerful applications of proteomics is to highlight proteins and pathways that underpin specific phenotypes. This approach requires quantitative comparison of 2 or more similar phenotypes, such as wild type and mutant. For this type of analysis, it is essential that a method for at least relative quantitation is employed.
The development of quantitative methods for proteomics has been an intense area of research. The three most common types of quantitative method are fluorescence labelling of 2D-gels, isotopic labelling and ‘label free’ proteomic analysis.
Gel comparison techniques
The great power of 2D gel electrophoresis lies in its ability to produce a high resolution analogue separation in two dimensions, a benefit not shared by any other protein separation technology (Gorg et al. Reference Gorg, Weiss and Dunn2004). This can be exploited in a quantitative fashion by using densitometry on the resulting gel spots. Gels can be stained (using the traditional Coomassie blue or, for increased sensitivity, silver staining or Sypro Orange can be used), scanned, and spot patterns matched by overlaying the gel images on top of each other. Modern 2D gel analysis software incorporates sophisticated warping algorithms that compensate for the small variations in spot pattern resulting from the gel microenvironment during separation. This allows multiple replicate images to be matched, facilitating inter-gel normalisation of spot intensities and relative quantitation. The technique, however, remains limited by the reproducibility of gels. To compensate for this limitation, Difference Gel Electrophoresis (DiGE) was implemented (Alban et al. Reference Alban, David, Bjorkesten, Andersson, Sloge, Lewis and Currie2003). This technique relies on labelling of three samples: control, treatment and a pooled sample derived from every replicate, with mass and charge-matched fluorophors that fluoresce under laser excitation with different colours – red, green and blue. These three samples are then mixed together and run on the same gel, which eliminates any gel-to-gel variation. Scanning followed by densitometry using a filter allowing only light of the appropriate wavelength for each sample to pass through provides a quantitative measure of the differences between control and treatment in a particular replicate set, and the pooled standard allows matching across gels more easily due to the presence of identical spots on each. Two different label types are currently available, a lysine labelling ‘minimal’ reagent for abundance samples, which labels only 1% of protein, and a cysteine-labelling saturation labelling reagent for low concentration samples (Kantawong et al. Reference Kantawong, Burchmore, Gadegaard, Oreffo and Dalby2008; McNamara et al. Reference McNamara, Dalby, Riehle and Burchmore2010).
The gel-based quantitative methods are unique in that quantitation is performed prior to extraction and identification of the proteins. This has a significant benefit – only differences which are significant and reproducible can be selected for characterization. The major limitation to this approach is that despite the high resolution separation available from a 2D gel, allowing several thousand protein spots to be visualised simultaneously, this is still a small fraction of the actual protein diversity present. This means that when a spot is excised and digested, it is common, indeed almost inevitable, that multiple proteins will be detected from each spot. This makes it difficult to ensure that a particular protein is responsible for an observed change in abundance, and validation is essential to confirm quantitation of a given protein.
Isotope Coded Affinity Tagging (ICAT)
ICAT is a saturation labelling technique that involves labelling proteins with two different mass tags, a ‘heavy’ and a ‘light’ tag at cysteine residues (Gygi et al. Reference Gygi, Rist, Gerber, Turecek, Gelb and Aebersold1999, Reference Gygi, Rist, Griffin, Eng and Aebersold2002). The heavy tag is isotopically differentiated from the light tag by 8 Da (original reagent) or 9 Da (latest generation cICAT reagent).
The latest generation cICAT reagent consists of four parts: a reactive group that binds to the protein, the aliphatic carbon chain tag itself (either 9×C12 or 9×C13), an acid-cleavable linker and a biotin tag which allows purification. Samples are initially reduced to expose all cysteine residues to chemical modification, followed by labelling with either the light or the heavy tag and then digestion. The two samples are then combined and purified away from excess reagent using cation exchange chromatography. A further stage of affinity chromatography using a streptavidin column is required to trap only those peptides labelled with biotin, which is then cleaved off with addition of concentrated TFA. The final mixture of cysteine-containing peptides is separated using shotgun 2-D liquid chromatography and analysed using MS. An algorithm searches the resulting data for peaks in the peptide MS spectra differing by 9 Da. Obtaining the area of each peak allows relative quantitation between control and test samples to be performed.
There are some limitations to the ICAT method. Approximately 8% of proteins contain no cysteine residues, and therefore will never be seen in a conventional ICAT experiment (Miseta and Csutora, Reference Miseta and Csutora2000). Additionally, because there are only two reagents, comparison of more than two samples can only be achieved by successive runs against a standard, which can introduce significant experimental error. Finally, due to the rarity of cysteine residues, only a few peptides are labelled in most proteins, which results in reduced confidence of quantitative analysis.
O18 labelling
The tryptic digestion of peptides is a hydrolysis reaction, requiring the incorporation of water molecules. The use of O18-labelled water in the digestion buffer will result in the incorporation of a single O18-labelled carboxyl group at the carboxyl end of each tryptic peptide (Schnolzer et al. Reference Schnolzer, Jedrzejewski and Lehmann1996; Yao et al. Reference Yao, Afonso and Fenselau2003). Thus a control sample may be digested with normal water and a test sample may be digested in the presence of O18-labelled water, resulting in peptides differing in mass by 2 Da. These may be quantified in the same manner as iCAT peptides. The main drawback to this type of quantitation is that the labelled peptides will occur at the same point as +2 and subsequent isotopic peaks of the unlabelled peptide. This significantly limits the dynamic range of the technique, although attempts have been made to improve quantitation through the use of isotope matching software (Johnson and Muddiman, Reference Johnson and Muddiman2004).
Dimethyl labelling
Dimethyl labelling was developed by Chen (Hsu et al. Reference Hsu, Huang, Chow and Chen2003, Reference Hsu, Huang and Chen2006) and is a non-isobaric protein and peptide labelling methodology based on the double methylation of lysine residues using either formaldehyde or deuterium-labelled formaldehyde. This method has a number of distinct advantages over other labelling methodologies, primarily that it is comparatively inexpensive and very rapid. Its low cost and high availability are indeed great benefits since a large excess of reagent can be used to improve labelling efficiency.
Isotope tagging for relative and absolute quantitation (iTRAQ)
The protocol for iTRAQ (Ross et al. Reference Ross, Huang, Marchese, Williamson, Parker, Hattan, Khainovski, Pillai, Dey, Daniels, Purkayastha, Juhasz, Martin, Bartlet-Jones, He, Jacobson and Pappin2004; Aggarwal et al. Reference Aggarwal, Choe and Lee2006) labelling is similar to that of ICAT. Samples to be quantified are reduced and alkylated (to prevent opportunistic chemical modification, such as oxidation of cysteine residues, and to prevent labelling of cysteine residues). They are then digested and labelled with the iTRAQ reagents. The nature of the label itself is significantly different: while the ICAT reagent targets cysteine residues, the iTRAQ tag reacts with the amino terminus of peptides and lysine side chains. Additionally, rather than the two different tags available in ICAT, four different iTRAQ reagents are available with masses of 114, 115, 116 and 117 and a recently available kit raises this number to eight, with a slight increase in the mass of the tag from 144 to 304. When attached to the parent peptide, iTRAQ tags are isobaric (possessing identical masses) and only a single peak is observed in each mass spectrum. Quantitation is observable only in MS/MS spectra, where the tags readily cleave, and the fragment ions appear in an unpopulated region in the MS/MS trace. By comparing the intensities of the 114, 115, 116 and 117 Da peaks, it is possible to obtain relative quantitation of peptides from multiple different samples. To obtain absolute quantitation, a standard with known abundance must be labelled with one of the tags and added to the mixture. This then serves as a calibrant for the remaining peaks. Drawbacks to the methodology are that, with the standard protocol, labelling is performed at a relatively late stage in the sample preparation process – after reduction, alkylation and digestion – and therefore significant experimental error may be introduced by the manipulations at each stage. Additionally, generation of intense reporter ions for quantitation along with high quality sequence data for identification of a peptide is difficult to achieve.
Stable isotope labelling with amino acids in cell culture (SILAC)
The technique of SILAC is based on the addition of isotopically labelled amino acids to culture medium (Ong et al. Reference Ong, Blagoev, Kratchmarova, Kristensen, Steen, Pandey and Mann2002). Cells are grown in medium containing C13- and/or N15-labelled amino acids (commonly lysine or arginine). Carbon starvation is sometimes employed to assist uptake of labelled amino acids. These labelled amino acids are taken up and incorporated into cellular proteins. Once cells are lysed, the lysates mixed together, and analysed by MS, it is possible to observe the relative abundances of a given labelled and unlabelled peptide or protein pair in terms of the spectral abundance of their individual mass peaks. The principal drawback of SILAC is that the technique works only with cells that can be grown on medium containing the labelled amino acid and that are auxotrophic for the relevant amino acid. The most commonly used amino acids for SILAC labelling are lysine and arginine, based on the principle that after a tryptic digestion every peptide (excepting the C terminal peptide) can be used for quantitation. Other amino acids suitable for SILAC labelling include leucine (which was used in the original report of the technique, Ong et al. Reference Ong, Blagoev, Kratchmarova, Kristensen, Steen, Pandey and Mann2002), and labelled methionine and tyrosine are commercially available. Lysine is commonly used in 12C6 and 13C6 versions for a mass shift of 6, but can be supplemented by 14N2 and 15N2 labels for a total mass shift of 8. Triplet labels can be implemented with the use of labelled arginine, as the 6 and 10 Da mass shifts between 12C614N4 arginine, 13C614N4 arginine and 13C615N4 arginine can be clearly visualised on most MS instruments.
While in vitro protein-tagging approaches such as DiGE, ICAT and iTRAQ can be applied to any proteome of interest, metabolic labelling approaches such as SILAC involve in vivo labelling. While this is a potential advantage because the introduction of bias during sample preparation is avoided, it does require that the material can be efficiently labelled. For many biological systems, including many parasites, culture is difficult under axenic, defined conditions and it can be difficult to generate labelled samples that have grown under physiologically relevant conditions. SILAC studies can thus necessitate significant culture adaptation before efficient labelling is achieved.
Direct quantitation by comparison of ion abundance (‘label-free quantitation’)
Both DeCyder MS (GE Healthcare), MSight (Palagi et al. Reference Palagi, Walther, Quadroni, Catherinet, Burgess, Zimmermann-Ivol, Sanchez, Binz, Hochstrasser and Appel2005, Reference Palagi, Muller, Walther and Lisacek2011) and Progenesis LC/MS (Nonlinear Dynamics) are developments of comparison software for 2-dimensional gel electrophoresis. DeCyder MS is based on the DeCyder software for DiGE analysis (GE Healthcare) and relies on the generation of 2-dimensional maps of peptide intensity, where mass is plotted against time, with ion abundance displayed as spot intensity. Once generated, these ‘pseudogel’ images can be overlaid and compared. Peaks are detected, and can be de-isotoped, deconvoluted and their charge state can be determined. After this stage, runs are overlaid and warped to provide peak matches. Comparison statistics are performed on the matched peaks, with rigour increasing with the number of replicated runs. MSight is public domain software based on Melanie (Appel et al. Reference Appel, Hochstrasser, Funk, Vargas, Pellegrini, Muller and Scherrer1991), which, like DeCyder, is designed for comparison of 2D gels. MSight is capable of the same kind of pseudogel generation and matching but lacks the peak processing facilities of DeCyder MS. Progenesis LC/MS is recently developed software produced by NonLinear Dynamics. It is a development of their SameSpot algorithm for 2-dimensional gel electrophoresis analysis. It provides built-in statistical analysis, including principle component analysis and clustering of the quantitative differences observed.
Unlike the chemical labelling techniques described above, where quantitation is performed in a single separation, direct MS quantitation relies on the comparison of mass spectrometry data, normally the different intensities of peaks between runs. It is therefore entirely reliant on the reasonable reproducibility of the mass chromatograms, and variation in processing between sample can have significant deleterious effects on the statistics.
The choice of an appropriate separation and quantitation approach depends upon the nature of the proteome under study, the type of questions asked and the instrumentation available. While high resolution protein and peptide separation is essential for a true proteomic analysis, more targeted questions may benefit from specific protein enrichment or fractionation approaches.
PROTEOMIC ANALYSIS APPLIED TO PARASITES
Proteomics is an approach that has developed rapidly as instrumentation and databases have improved to make specific questions more tractable. Proteomic technologies have generic applicability across biological research, and many proteomic practitioners are focused on technical aspects. Thus it is essential for researchers from specific fields, such as parasitologists, to have an initial understanding of what is achievable using proteomic approaches. General considerations have recently been reviewed from a pragmatic perspective (Mallick and Kuster, Reference Mallick and Kuster2010). The aim of the following section is to illustrate some of the ways in which proteomic technologies can be applied in parasitology.
Affinity purification of proteins
Despite increasing emphasis on target-based drug screening, many potential anti-infective drugs are identified through phenotype screens. Elucidation of mode of action for such compounds is now generally a prerequisite for their approval as drugs. Thus it is important to identify the molecular targets of drugs that show desirable activities, not least because this information can be fed back into target-based screening pipelines. Proteomic technologies are well suited to the identification of putative drug targets, since the requirement is for a non-hypothesis-driven screen. The most direct approach to drug target screening is by chemical proteomics, in which a drug of interest is immobilised and used to enrich interacting proteins by affinity. After washes of appropriate stringency, bound proteins can be eluted and characterized by mass spectrometry. For example, this approach has recently been exploited to identify potential targets for salicylidene acylhydrazine compounds that are active against a variety of bacterial pathogens (Wang et al. Reference Wang, Zetterstrom, Gabrielsen, Beckham, Tree, Macdonald, Byron, Mitchell, Gally, Herzyk, Mahajan, Uvell, Burchmore, Smith, Elofsson and Roe2011). These compounds act as virulence-blocking agents and exposure of pathogenic E.coli O157 to these drugs results in strong transcriptional repression of components of the type III secretion system, potentially implicating the important virulence system as a target for salicylidene acylhydrazine (Tree et al. Reference Tree, Wang, McInally, Mahajan, Layton, Houghton, Elofsson, Stevens, Gally and Roe2009). An Affigel-coupled salicylidene acylhydrazine analogue was synthesised, with linker addition sited to avoid groups that were previously found to be important for activity. This medium was used to generate an affinity chromatography column, over which E.coli lysate was passed (Fig. 3). The column was washed extensively, until protein could no longer be detected in the washes. Proteins were then eluted from the column by addition of free compound, to promote competitive dissociation of specifically bound proteins. The column was subsequently stripped by acid elution and the two eluates were fractionated by SDS-PAGE. Gel staining revealed less than 20 detectable protein species of varying abundance in the specifically eluted fraction. Some of these, in particular some proteins of relatively high abundance, were also observed in the non-specific eluate that derived from column stripping, suggesting that these species may not interact specifically with the salicylidene acylhydrazine moiety of the affinity matrix. Protein bands were excised from the gel, subjected to in-gel digest and the resulting peptides analysed by LC-MS. In total, 16 E. coli proteins were confidently identified by Mascot searching. Interestingly, several of these proteins play known or putative roles in the biogenesis or function of the type III secretion system. To test individually the ability of each identified protein to interact with salicylidene acylhydrazine compounds, the encoding gene for each was selected for cloning and recombinant expression. This was achieved for 7 of the proteins of interest and the potential for each protein to interact with drug was tested by far Western blotting. Three of the 7 proteins identified were found to interact with labelled drug, thus validating some of the putative targets identified by the chemical proteomics screen (Wang et al. Reference Wang, Zetterstrom, Gabrielsen, Beckham, Tree, Macdonald, Byron, Mitchell, Gally, Herzyk, Mahajan, Uvell, Burchmore, Smith, Elofsson and Roe2011).

Fig. 3. Affinity chromatography to identify drug targets. Affinity chromatography enables the selection of a sub-proteome based on ligand binding.
Chemical proteomics is at the interface between synthetic chemistry and biochemistry, and has the potential to make a significant contribution to the development of new drugs (Bantscheff et al. Reference Bantscheff, Scholten and Heck2009). The mechanisms of action for many anti-parasitic drugs are poorly understood, and a chemical proteomics approaches might help to elucidate some of these mechanisms, helping to explain how drug resistance can arise and might be averted, and also facilitating the rational optimisation of drugs that are invaluable but which have undesirable side effects. As with most proteomic approaches, it is important to appreciate that affinity enrichment of interacting proteins is subject to significant background effects. Proteins will bind non-specifically to the affinity matrix, and may also bind specifically to chromatography resin or to the linker (Trinkle-Mulcahy et al. Reference Trinkle-Mulcahy, Boulon, Lam, Urcia, Boisvert, Vandermoere, Morrice, Swift, Rothbauer, Leonhardt and Lamond2008). Conversely, the covalent coupling of drug to linker may occlude or ablate functional groups or generate new groups that have different activities. Thus it is critical that targeted approaches are used to validate putative drug:target interactions.
Furthermore, the structural conservation that is evident within and between protein families means that most drugs do not bind to a single target. While multi-target effects can be advantageous if they increase drug potency or limit the selection of resistance, they are also responsible for significant side effects (sometimes considered as ‘off-target’ effects). Some drug classes show frankly promiscuous interactions that can greatly complicate elucidation of mechanisms of action. Chemical proteomics has the potential to identify multiple protein targets in a single step, but the validation of multiple putative targets is critical but time consuming.
Despite the non-hypothesis-driven nature of a chemical proteomics screen, it must be acknowledged that the approach is inherently biased towards abundant soluble proteins. If membrane proteins are of likely interest (and primary drug targets are receptors or transporters) it is necessary to develop appropriate lysis conditions that will solubilise these proteins and enable them to interact with the immobilised ligand. The possibility that the presence of detergents and chaotropes might alter binding characteristics further underlines the criticality of targeted validation approaches.
Characterization of affinity purified protein complexes
Affinity purification is a powerful tool for proteomic analysis enabling proteins to be isolated with a high degree of specificity. Affinity purification approaches can permit the characterization of proteins that physically interact in cells and have been exploited to elucidate the composition and stoichiometry of structures of significant complexity (Heck and van den Heuvel, Reference Heck and van den Heuvel2004; DeGrasse et al. Reference DeGrasse, Chait, Field and Rout2008). The use of antibodies to isolate specific proteins, together with associated proteins can enable the mapping of protein:protein interactions and the definition of protein complexes. However, studies of this type can be confounded by the inclusion of proteins that are not physiological components of the complex under study. When cells are lysed to enable affinity purification, artifactual protein:protein interactions can result. Furthermore, unrelated proteins can associate with the enriched complex or with the chromatography matrix. Although this background contamination with irrelevant protein can be reduced by stringent washing prior to elution, washing can also promote the dissociation of physiologically relevant interactions. Comparison between eluates from affinity columns and negative control columns that are charged with no affinity target, or with an irrelevant target, can go some way towards sorting biologically relevant interactors from artifacts. For example, sepharose beads, which are commonly used as an affinity chromatography matrix, have been shown to bind a wide variety of proteins, including many abundant cytoskeletal proteins (Trinkle-Mulcahy et al. Reference Trinkle-Mulcahy, Boulon, Lam, Urcia, Boisvert, Vandermoere, Morrice, Swift, Rothbauer, Leonhardt and Lamond2008). Quantitative proteomics approaches, particularly metabolic labelling strategies such as SILAC (stable isotope labelling with amino acids in cell culture), are particularly powerful ways to reveal proteins that are over-represented in affinity purified material compared with negative controls. Differential labelling approaches are costly and metabolic labelling strategies such as SILAC have yet to be developed for many non-model organisms, including most parasites.
Organellar proteomics
Subcellular fractionation represents an alternative strategy to focus proteomic analyses which can more readily be resolved into individual protein components. This approach exploits classical biochemical strategies, in particular isopycnic centrifugation, for the preparative fractionation of cells based on the buoyant density of their component organelles. This strategy exploits the intricate organisation of cells into organelles and can enable the generation of significantly enriched fractions in specific organelles. Although this procedure may require some optimisation when applied to specific cell types or organisms, efficient protocols have been developed and reported for many organisms, including parasites. Such approaches have been applied with particular effect to trypanosomes which have a complex ultrastructure despite a relatively simple genome. Various subcellular fractionation approaches have resulted in the description of sub-proteomes for trypanosome plasma membrane and cytoskeleton (Bridges et al. Reference Bridges, Pitt, Hanrahan, Brennan, Voorheis, Herzyk, de Koning and Burchmore2008), flagellum (Broadhead et al. Reference Broadhead, Dawe, Farr, Griffiths, Hart, Portman, Shaw, Ginger, Gaskell, Mckean and Gull2006), glycosome (Colasante et al. Reference Colasante, Ellis, Ruppert and Voncken2006), nucleus (Rout and Field, Reference Rout and Field2001) and mitochondrion (Panigrahi et al. Reference Panigrahi, Ogata, Zikova, Anupama, Dalley, Acestor, Myler and Stuart2009). Although each of these subcellular fractions have proven to be significantly contaminated with proteins that derive from other cellular compartments, the degree of enrichment obtained permits the identification of relatively rare proteins that would likely not have been detected in comparable proteomic analyses of unfractionated trypanosome proteomes (Jones et al. Reference Jones, Faldas, Foucher, Hunt, Tait, Wastling and Turner2006).
This is well illustrated by the characterization of the bloodstream form Trypanosoma brucei plasma membrane proteome (Bridges et al. Reference Bridges, Pitt, Hanrahan, Brennan, Voorheis, Herzyk, de Koning and Burchmore2008). As for all protozoan parasites, the surface of the trypanosome is the interface with the host and proteins associated with the surface are important to host:parasite interactions. Membrane proteins include those that are peripheral to the lipid bilayer that comprises the membrane and those that are integral to the lipid bilayer. Integral membrane proteins are soluble in the lipid bilayer and are thus relatively hydrophobic. They are also generally of relatively low abundance, as they are constrained in the 2-dimensional matrix of the membrane, unlike globular proteins which occupy the 3-dimensional space within the cell and its organelles. The low abundance of integral membrane proteins means that they are under-represented in global proteomic analyses which are dominated by more abundant proteins. Hydrophobicity compounds this problem because hydrophobic proteins are poorly soluble and may thus be relatively refractory to enzymatic digest and, if digested, may yield hydrophobic peptides that ionise poorly in mass spectrometry. One approach to enhance coverage of a membrane proteome is to isolate membranes, thus greatly reducing the distracting presence of abundant non-membrane proteins. Combined with membrane protein-optimised sample preparation and mass spectrometry, it is possible to increase the coverage of integral membrane proteins significantly.
The enrichment of trypanosome plasma membranes takes advantage of the intimate association between the plasma membrane and the subtending microtubule-based cytoskeleton (Voorheis et al. Reference Voorheis, Gale, Owen and Edwards1979). Bloodstream form trypanosomes were lysed by hypotonic stress and washed by centrifugation to produce a lysate in which plasma membrane sheets remained associated with the cytoskeleton. The specific buoyant density of these plasma membrane-cytoskeleton fragments enabled their enrichment by sucrose density gradient centrifugation. The enriched plasma membrane fraction thus obtained was resolved by a variety of proteomic approaches, ultimately leading to the identification of some 1200 gene products. Parallel subcellular fractionation to enrich membrane-free trypanosome cytoskeletons enabled the characterization of this sub-proteome and the subsequent subtraction of cytoskeletal proteins from the plasma membrane proteome. This process left some 600 proteins that were identified in the plasma membrane fraction but not in the cytoskeleton fraction. Relatively rare hydrophobic proteins (predicted respectively by codon usage and hydropathy analyses) were over-represented in this subset, suggesting that it did indeed include membrane proteins (Bridges et al. Reference Bridges, Pitt, Hanrahan, Brennan, Voorheis, Herzyk, de Koning and Burchmore2008). Some 10% of the proteins assigned to the plasma membrane fraction were apparent integral membrane proteins, with 5 or more hydrophobic integral membrane domains. These included many nutrient transporters, channels, porins and hypothetical proteins. There were many other proteins with one or more hydrophobic integral membrane domains, notably a large group of receptor adenylate cyclases, that have putative signalling roles. The identification of such a large cohort of membrane proteins demonstrates the power of the targeted sub-proteomic approach taken because proteomic analyses of unfractionated trypanosomes result in the identification of few putative membrane proteins.
Bioinformatic analysis of the trypanosome genome sequence suggests that some 350 proteins contain 5 or more integral membrane domains. Fewer than 20% of this number were observed in proteomic analysis of the bloodstream form trypanosome plasma membrane fraction. At face value, this is a low proportion but it should be considered that the trypanosome has many organellar membranes that will be populated with specific membrane proteins. Furthermore, many of the encoded membrane proteins may be expressed under particular environmental conditions or may be expressed exclusively in the procyclic stage of the parasite. Thus the membrane proteins identified by this proteomic approach are likely to be a very significant proportion of those actually expressed in the bloodstream from trypanosome plasma membrane. Some plasma membrane proteins are clearly missing from this proteomic characterization, most notably the trypanosome hexose transporter, THT1, which provides the cell with essential glucose (Barrett et al. Reference Barrett, Tetaud, Seyfang, Bringaud and Baltz1998).
The mechanism by which the plasma membrane fraction was generated would seem to leave little possibility for significant contamination with membrane proteins from other compartments. However, there is clear possibility for contamination with soluble or peripheral membrane proteins that are not bona fide citizens of the plasma membrane. Indeed, many of the proteins identified in this fraction have functions elsewhere in the cell and have likely become associated with the plasma membrane fraction during isolation. From the results of the proteomic analysis alone, it is impossible to determine which of these proteins is a simple contaminant and which might have an unexpected association with the trypanosome plasma membrane. Targeted studies are required to investigate the subcellular localisation of these proteins.
Serological proteomics
Many components of a parasite proteome may present attractive drug targets or may account for phenotypes such as virulence. A restricted subset of the total proteome is recognised by the host immune response and may thus represent vaccine targets. Identification of antigenic proteins may also help to dissect host:parasite interactions. Proteomic approaches can be applied to identify antigenic proteins, by exploiting Western blotting to localise proteins that are recognised by sera from infected or immune hosts. For this approach to be effective, intact proteins must be separated with maximum resolution, an ideal application for 2-D electrophoresis. Potential antigens, whether in the form of an unfractionated lysate or a specific fraction, are separated in parallel on two similar 2-dimensional gels. One of the resulting gels is strained to generate a spot map that represents the total proteome. The other is blotted and probed with host serum to generate an antigen spot map. Cross-reference between the two spot maps can enable the localisation of protein spots that are antigenic, and these can then be excised for subsequent identification.
For example, proteins extracted from nematode Teladorsagia circumcincta, an important parasite of sheep, were separated by 2-DE and antigenic proteins highlighted by Western blot with serum from infected sheep (Murphy et al. Reference Murphy, Eckersall, Bishop, Pettit, Huntley, Burchmore and Stear2010). Several potential antigens were identified by this approach. In another similar example, Schistosoma haematobium antigens were identified by screening 2D Western blots with patient serum (Mutapi et al. Reference Mutapi, Burchmore, Mduluza, Midzi, Turner and Maizels2008, Reference Mutapi, Bourke, Harcus, Midzi, Mduluza, Michael, Turner, Burchmore and Maizels2010). The serological proteomics approach has great potential to shed light on molecular interactions between parasite and immune system but has largely been exploited to date to investigate abundant antigens. There is great potential to use this approach to screen for proteins that are much less abundant but highly antigenic and to search for antigens that are potentially protective rather than just highly antigenic proteins. Fractionation of parasite proteomes could enhance proteomic coverage to include less abundant proteins while differential screening of Western blots with sera from naïve, immune and infected hosts could highlight specific proteins whose immune recognition is correlated with protection.
Comparative proteomic analysis
One of the most powerful applications of proteomic technologies is as a tool to screen, without preconception, for differences in protein expression. Such approaches can be exploited to highlight molecular changes that accompany a specific phenotype. In the context of parasitology, comparative proteomic analyses have a myriad of applications to deconvolute phenotypes that have relevance to disease. For example, comparative proteomic approaches have been applied to compare different life cycle stages (Walker et al. Reference Walker, Vasquez, Gomez, Drummelsmith, Burchmore, Girard and Ouellette2006; Rosenzweig et al. Reference Rosenzweig, Smith, Opperdoes, Stern, Olafson and Zilberstein2008; Tarun et al. Reference Tarun, Peng, Dumpit, Ogata, Silva-Rivera, Camargo, Daly, Bergman and Kappe2008; Paape et al. Reference Paape, Lippuner, Schmid, Ackermann, Barrios-Llerena, Zimny-Arndt, Brinkmann, Arndt, Pleissner, Jungblut and Aebischer2008), to reveal changes in drug resistant parasites (for example, Foucher et al. Reference Foucher, McIntosh, Douce, Wastling, Tait and Turner2006; Briolant et al. Reference Briolant, Almeras, Belghazi, Boucomont-Chapeaublanc, Wurtz, Fontaine, Granjeaud, Fusai, Rogier and Pradines2010) and to address host responses to parasitism (Nelson et al. Reference Nelson, Jones, Carmen, Sinai, Burchmore and Wastling2008). These studies, exploiting a variety of the quantitative approaches described earlier, report on only a minority of the total proteome but reveal expression data for a cross-section, often enabling specific pathways to be implicated and providing clues that direct subsequent more targeted studies.
A recent study used a comparative 2D gel-based approach to investigate proteomic changes in an attenuated Leishmania infantum line (Daneshvar et al. Reference Daneshvar, Wyllie, Phillips, Hagan and Burchmore2012). Expression of more than 2,000 protein species was assessed by difference gel electrophoresis (DiGE) resulting in the identification of 18 proteins that showed significant and reproducibly altered expression (P=<0·01; n=4) of greater than 2-fold (Fig. 4). Several of the modulated proteins were known to be involved in redox control and, when response to oxidative stress was assessed, the attenuated line was shown to be more susceptible.

Fig. 4. DiGE analysis of attenuated Leishmania infantum. Arrows indicate selected proteins that showed significant and reproducible modulation of expression in attenuated Leishmania, compared with wild type Leishmania. Inset graphs indicate average fold change in expression, with individual lines for replicate analyses (n=4). Inset diagram illustrates the key role of tryparedoxin peroxidase in Leishmania thiol-redox control.
Proteomic analysis of parasites has the potential to highlight key molecules in clinically relevant phenotypes such as virulence and drug resistance. and to identify potential drug and vaccine targets. The availability of genome sequence information for an increasing range of parasites of human and veterinary relevance facilitates the application of many generic proteomic approaches to parasites. In addition, the sensitivity and speed of the instrumentation available is constantly improving, allowing deeper analysis of parasitic systems. Finally, the diverse methodologies available for accurate quantitation of parasitic proteins allow meaningful differences to be obtained from most experimental designs. If the right questions are asked, proteomics will make a significant contribution to parasitology.