INTRODUCTION
Sri Lanka is attempting to eliminate malaria from its borders by the end of 2014. Potential for malaria transmission exists throughout the year in Sri Lanka, with 11·7 million people at risk of infection, mostly due to Plasmodium vivax. The incidence of malaria in Sri Lanka has drastically declined over the past decade, with 264 549 confirmed malaria cases in 1999 but only 23 indigenous cases reported in 2012 (WHO, 2012; Anti-Malaria Campaign, Sri Lanka; Fig. 1). All reported cases were examined either microscopically or by using rapid diagnostic tests (RDT) by the National Malaria Control Programme of the Ministry of Health in Sri Lanka. The use of mobile malaria clinics, distribution and use of long-lasting insecticide nets (LLINs) along with aggressive indoor residual spraying for vector control produced excellent results (World Malaria Report, 2010). The current approach to elimination is based on strengthened surveillance, early reporting, case investigation and case management with radical cure (WHO, 2012). Since 2008, artemisinin-based combination therapy was introduced for the treatment of uncomplicated Plasmodium falciparum infections and surveillance through active case detection was intensified targeting elimination of malaria from Sri Lanka (WHO Country Reports, 2008).

Fig. 1. Total confirmed malaria infections in Sri Lanka from 1995 to 2012.
The operational challenges of elimination and prevention of resurgence of malaria are numerous. Plasmodium vivax is more challenging to eliminate than P. falciparum due to more asymptomatic and subclinical infections, infections at lower parasite densities making detection more difficult, the ability of the parasite cycle in the vector to exist at lower temperatures, and the existence of hypnozoites, the dormant liver stage that causes relapses (Abeyasinghe et al. Reference Abeyasinghe, Galappaththy, Smith Gueye, Kahn and Feachem2012). Another major challenge to sustaining elimination is addressing the potential reintroduction of cases, either via border areas or from migrant populations (Global Malaria Action Plan, 2011), as most parasites among neighbouring areas are exchanged through human migrations. The geographic isolation of islands can limit malaria importation and may make control easier (Rebaudet et al. Reference Rebaudet, Bogreau, Silaï, Lepère, Bertaux, Pradines, Delmont, Gautret, Parola and Rogier2010).
Sri Lanka has the advantage of being an island and would require stringent surveillance measures at ports of entry to the country to prevent reintroduction of malaria, particularly cases that would lead to re-establishment of local transmission. If the receptivity and vulnerability to malaria in an area is minimal, the probability of reintroduction is minimal. However, the risk varies across regions and can change seasonally or with other factors such as population movement and development projects (such as irrigation projects, mining and forest clearing) which create favourable conditions for vectors and increase human-vector contact (Global Malaria Action Plan, 2011). With the dawn of a new era of rapid development and growth in Sri Lanka following the end of a 3 decade long civil war in 2009, population movement as well as development projects have dramatically increased. The risk of importation of malaria is greatly increased with the boom in tourism, resumption of ferry services to and from southern India as well as other smaller boat traffic between the countries in the region (WHO, 2012). The role of irregular migration in the form of human trafficking as an important pathway to re-introduction of malaria has also been highlighted recently (Wickramage et al. Reference Wickramage, Premaratne, Peiris and Mosca2013).
Complete interruption of local transmission of malaria will require effective tools and greatly strengthened monitoring and surveillance programmes (WHO, 2008). A re-orientation of control activities, moving away from a population-based coverage of interventions, to one based on a programme of effective surveillance and response is required. Sustained efforts will be required to prevent the resurgence of malaria from where it is eliminated (Mendis et al. Reference Mendis, Rietveld, Warsame, Bosman, Greenwood and Wernsdorfer2009). Failure to maintain the reduced levels of malaria thus achieved will lead to rebound epidemics with disastrous results, as evidenced during the 1967–68 periods in Sri Lanka when the malaria eradication programme that was embarked upon at the time had to be abandoned.
Genotyping of P. vivax using microsatellite markers has proven to be a useful tool in identifying short-term outbreaks as well as in determining the ancestry of parasite isolates, enabling differentiation between Asian and African isolates (Gunawardena et al. Reference Gunawardena, Karunaweera, Ferreira, Phone-Kyaw, Pollack, Alifrangis, Rajakaruna, Konradsen, Amerasinghe, Schousboe, Galappaththy, Abeyasinghe, Hartl and Wirth2010). With Sri Lanka making tremendous progress towards achieving its target of malaria elimination by 2014, genetic surveillance may be useful in evaluating the effectiveness of the control strategies implemented. Population genetic theory predicts that in a small population, random genetic drift, inbreeding and the decreased efficiency for natural selection will lead to a loss of genetic variation (Frankham et al. Reference Frankham, Ballou and Briscoe2003). Given the drastic reduction in malaria transmission in recent years, the Sri Lankan P. vivax population presents a good opportunity to look for signs of a bottleneck in genetic data.
MATERIALS AND METHODS
In this paper we have re-analysed in whole the data of Sri Lankan parasite isolates that have been described previously and published in Karunaweera et al. (Reference Karunaweera, Ferreira, Hartl and Wirth2007, Reference Karunaweera, Ferreira, Munasinghe, Barnwell, Collins, King, Kawamoto, Hartl and Wirth2008) and Gunawardena et al. (Reference Gunawardena, Karunaweera, Ferreira, Phone-Kyaw, Pollack, Alifrangis, Rajakaruna, Konradsen, Amerasinghe, Schousboe, Galappaththy, Abeyasinghe, Hartl and Wirth2010).
Field isolates
We genotyped 185 P. vivax field isolates collected from 13 districts within Sri Lanka (Fig. 2) between 2003 and 2007. In order to minimize effects of sampling effort, temporal trends for statistics of interest were determined by pooling samples from 2003–4, 2005 and 2006–7; for evaluating geographical differentiation and structure, samples were considered according to provinces. Venous blood samples were collected from these patients after informed consent and DNA templates for PCR amplification were isolated using either the standard phenol-chloroform method or the Nucleon genomic DNA extraction kit (Tepnel Life Sciences, Manchester, UK). Due to the limited availability of genomic DNA in these samples, the extracted DNA was further subjected to whole genome amplification (WGA) using a REPLI-g Mini-kit (Qiagen, Valencia, CA). The study protocol was approved by the Human Subjects Committee of the Harvard School of Public Health (#P10299-111/0209GENE) and the Ethical Review Committee of the Faculty of Medicine, University of Colombo (EC/08/092).

Fig. 2. Map of Sri Lanka showing the districts from where the 185 field isolates were collected. N=sample numbers obtained from each location. Shown inset is a map of Sri Lanka demarcating the nine provinces.
Microsatellite typing of P. vivax
We amplified 14 highly polymorphic microsatellite markers using previously validated primer sets and PCR protocols (Karunaweera et al. Reference Karunaweera, Ferreira, Hartl and Wirth2007, Reference Karunaweera, Ferreira, Munasinghe, Barnwell, Collins, King, Kawamoto, Hartl and Wirth2008). Length variation of labelled PCR products was measured on an ABI PRISM 3730XL DNA Analyzer (Applied Biosystems, Foster City, CA) using ABI GS500LIZ internal size standards and the GENESCAN, GENOTYPER and GENEMAPPER software (Applied Biosystems).
Genetic diversity and population differentiation
The single or predominant allele at each locus was considered for computing allele frequencies (Anderson et al. Reference Anderson, Haubold, Williams, Estrada-Franco, Richardson, Mollinedo, Bockarie, Mokili, Mharakurwa, French, Whitworth, Velez, Brockman, Nosten, Ferreira and Day2000). Because blood-stage malaria parasites are haploid, each peak on the resulting pherogram was considered a single clone of parasite. The presence of additional alleles, having a peak of at least one third the height of the predominant allele, at a particular locus was considered as multiple-clone infections (MCI) in the same isolate (Anderson et al. Reference Anderson, Su, Bockarie, Lagog and Day1999; Imwong et al. Reference Imwong, Nair, Pukrittayakamee, Sudimack, Williams, Mayxay, Newton, Kim, Nandy, Osorio, Carlton, White, Day and Anderson2007a ). The eBurst v3 software (Feil et al. Reference Feil, Li, Aanensen, Hanage and Spratt2004) was used to search for nearly identical multilocus haplotypes (those differing by a single locus; Cooper et al. Reference Cooper, Amos, Hoffman and Rubinsztein1996). Genotypic richness index (R), was used to examine changes in the distribution of genotypes temporally and spatially. R measures the proportion of unique genotypes present in the samples and is estimated as R = (G−1)/(N−1) where G is the number of distinct genotypes and N is the sample size (Dorken and Eckert, Reference Dorken and Eckert2001; Nkhoma et al. Reference Nkhoma, Nair, Al-Saai, Ashley, McGready, Phyo, Nosten and Anderson2012).
The genetic diversity of the parasite population was determined by calculating the virtual heterozygosity (H E ) at each locus using LIAN software 3.5 (Haubold and Hudson, Reference Haubold and Hudson2000). Virtual heterozygosity estimates the average probability that a pair of alleles randomly obtained from the population is different and was defined as H E = [n/(n−1)][1−Σp 2 i], with n being the number of isolates analysed and p the frequency of the i-th allele in the population.
Genetic differentiation between populations was estimated by pairwise fixation index (F ST ), with 10 000 permutations and significance based on a permutation process using Arlequin software 3.1 (Excoffier et al. Reference Excoffier, Laval and Schneider2006).
Linkage disequilibrium
The overall multilocus linkage disequilibrium (LD) analysis was evaluated using a standardized index of association (IS A ) in the parasite population according to spatial and temporal distribution and was defined as: IS A = (VD/VE−1)(r−1) with r being the number of loci analysed (Hudson, Reference Hudson1994). Analyses were performed using LIAN software version 3.5 (Haubold and Hudson, Reference Haubold and Hudson2000).
Effective population size and Bottleneck signatures
The effective population size (Ne) of P. vivax was estimated using the method of Anderson et al. (Reference Anderson, Haubold, Williams, Estrada-Franco, Richardson, Mollinedo, Bockarie, Mokili, Mharakurwa, French, Whitworth, Velez, Brockman, Nosten, Ferreira and Day2000). This measure of Ne uses the genetic diversity in the population and an estimated mutation rate (μ) for Plasmodium. Estimates were based on both the infinite allele model (IAM) and the step-wise mutation model (SMM). For SMM, Ne is given by Neμ = 1/8 {[1/(1-H E )]2–1} and for IAM, Neμ = He/4(1−H E ), where H E is the mean expected heterozygosity across all loci.
We used three methods to verify whether the population had suffered a reduction in size such as a bottleneck effect: (i) Mode shift test (Luikart et al. Reference Luikart, Allendorf, Cornuet and Sherwin1998); (ii) the heterozygote excess test implemented in Bottleneck 1.2.02 software (Piry et al. Reference Piry, Luikart and Cornuet1999) and (iii) M-ratio test (Garza and Williamson, Reference Garza and Williamson2001). Each of these tests requires a single temporal sample and is capable of detecting bottlenecks given the sample size and number of loci used in this study. In the first test we examined the allele frequency distribution for all loci combined looking for a ‘mode shift’ in the distribution (Luikart et al. Reference Luikart, Allendorf, Cornuet and Sherwin1998). This qualitative graphical test is based on the premise that a bottleneck causes a reduction in the relative abundance of rare alleles resulting in a mode shift in allele frequency distribution. In using Bottleneck (Piry et al. Reference Piry, Luikart and Cornuet1999), we tested for an excess of heterozygosity brought about by the loss of rare alleles following a population bottleneck. We assumed a two-phase mutational (TPM) model of 70% stepwise and 30% non-stepwise mutations and run 10 000 iterations. The third method we used to test for recent genetic bottleneck was the M-ratio test (Garza and Williamson, Reference Garza and Williamson2001). The M-ratio test is based on the premise that most allelic states of any microsatellite locus should be occupied, assuming a SMM and a robust population size. Because rare alleles are lost during a bottleneck, there tends to be an increase in the number of unoccupied allelic states, which can be quantified by expressing the ratio of the number of alleles observed in a single population sample (k) to the number of potential allelic states given the size range of alleles (r) observed in the sample.
RESULTS
Plasmodium vivax genotypes
All Sri Lankan parasite isolates were highly polymorphic and showed 184 unique haplotypes (genotypic richness R⩾0·97; Table 2), with the number of alleles per locus varying between 12 and 46 (mean = 21·57; Table 1, Supplementary Tables 1 and 2). Only a single haplotype was shared between isolates (SL138-SL139 collected from Puttalam district in 2005). These two isolates were from separate individuals of different ethnic origin, living approximately 10 km apart and collected on subsequent days. Closely related haplotypes (differing at a single locus) were seen between 5 pairs of Sri Lankan isolates; SL18-SL19 collected in 2003 from Kataragama, SL142-SL145 and SL149-SL150 collected in 2005 from Puttalam, and SL177-SL185 and SL173-SL179 collected from Trincomalee during an outbreak in 2007. Except for one of the above pairs which was collected on the same day from two different persons (SL 149-SL150), all others were collected from different individuals on different days ranging from 1–3·5 months apart. The outbreak samples from Trincomalee (n = 21) were collected just over a period of 6 months.
Table 1. Characterization of the 14 polymorphic P. vivax microsatellite loci (n = 185)

Table 2. Temporal trends in genetic parameters of P. vivax isolates from Sri Lanka

Multiple clone infections
The overall number of MCI was high (n = 127; 68·6%; Table 2). MCI in the 2003–4 period was almost 54%, increasing greatly in 2005 to 85% and then declining to 59% in the 2006–7 period. When we considered as evidence of multiple infections the presence of more than one peak for two or more loci, this same trend held true although the numbers of MCI reduced by 15–30% (Table 2). In the provinces too MCI were high (>70%) except at Uva. We further analysed for MCI in isolates obtained from Trincomalee (Eastern province) in 2005 and 2007. Almost all samples (19/20) in 2005 were found to be MCI compared with only 6/21 in 2007.
Virtual heterozygosity
The virtual heterozygosity (HE) values per locus for the entire Sri Lankan parasite population were high, ranging between 0·8015 and 0·9657 (mean = 0·8744; s.e.±0·0137; Table 1). A gradual decline in H E was observed from 2003–4 to 2006–7 (Table 2). High H E was seen even according to the provinces of distribution (Table 3). To prevent overestimation due to pooling of diverse samples, we determined H E at individual sites as well (range: 0·6447 at Puttalam to 0·8613 at Polonnaruwa; data not shown). The geographical and temporal distribution of alleles derived from genotyping 185 P. vivax isolates using 14 microsatellite markers is summarized in the Supplementary Tables 1 and 2.
Table 3. Genetic parameters of P. vivax isolates according to their provinces of distribution

Population differentiation
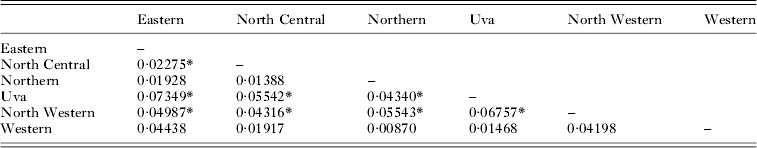
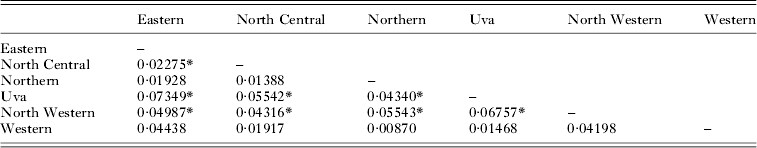
Genetic differentiation of P. vivax populations (FST) between 11 locations in 10 districts is shown in Table 4. Kataragama which belongs to the Monaragala district and situated at its southern border was considered separately. Three districts were excluded from the analysis due to sample size limitations. Low but significant differentiation between several districts (at P⩽0·0009 after Bonferroni multiple test correction) was confirmed with F ST estimates varying between 0·04 and 0·25 (Table 4). Analysis of genetic differentiation of P. vivax populations between six provinces (1 province with only a few parasite isolates was not considered), revealed significant levels of differentiation except in the Western province (Table 5). Further analysis was done to test if the years of sample collection had an effect on genetic differentiation. Plasmodium vivax parasite isolates obtained between 2003 and 2007 revealed significant temporal differentiation (even at P = 0·0001) and is shown in Table 6. Isolates collected from Trincomalee in 2005 (n = 20) and in 2007 (n = 21) revealed significant genetic differentiation as well (F ST = 0·09, P = 0·0001).
Table 4. Genetic differentiation (F ST) between P. vivax populations from 11 locations (10 districts) in Sri Lanka

* Significant at P⩽0·0009 after Bonferroni correction for multiple tests.
TRC, Trincomalee; ANP, Anuradhapura; PLN, Polonnaruwa; VVA, Vavuniya; BTC, Batticaloa; AMP, Ampara; MNG, Monaragala; KGL, Kurunegala; CMB, Colombo; PTM, Puttalam; KTM, Kataragama.
Table 5. Genetic differentiation (F ST) between P. vivax populations from six provinces in Sri Lanka
* Significant at P⩽0·0033 after Bonferroni correction for multiple tests.
Table 6. Genetic differentiation (F ST) between P. vivax populations obtained over 5 consecutive years in Sri Lanka

*Significant at P⩽0·005 after Bonferroni correction for multiple tests. **Significant even at P = 0·0001.
Linkage disequilibrium
Significant LD was seen for the whole population (IS A = 0·0279; P<0·001) as well as for subpopulations from 2003 to 2007: IS A = 0·02 (2003–4), 0·05 (2005), 0·14 (2006–7); P<0·01; Table 2). The association was extremely high in the 2007 subpopulation (samples from a known outbreak in Trincomalee) in comparison to the other subpopulations (2007, n = 21; IS A = 0·31), and these outbreak isolates from Trincomalee (Eastern province, Fig. 2) are genetically closer as revealed by previous analysis (Gunawardena et al. Reference Gunawardena, Karunaweera, Ferreira, Phone-Kyaw, Pollack, Alifrangis, Rajakaruna, Konradsen, Amerasinghe, Schousboe, Galappaththy, Abeyasinghe, Hartl and Wirth2010). Significant LD persisted even according to different provinces (Table 3). Since significant LD could be due to closely related infections sampled from the same location within a short span of time, we considered samples from the 2003–4 period with the omission of the cluster from Kataragama and the 2005 samples omitting clusters from Puttalam, Batticoloa and Monaragala. Significant LD still held true although the values were reduced considerably (IS A = 0·01; P = 0·002 for 2003–4 and IS A = 0·02; P<0·001).
Effective population size and bottleneck signatures
We estimated the effective population size (Ne) over the years (2003–4, 2005, 2006–7) based on the method of Anderson et al. (Reference Anderson, Haubold, Williams, Estrada-Franco, Richardson, Mollinedo, Bockarie, Mokili, Mharakurwa, French, Whitworth, Velez, Brockman, Nosten, Ferreira and Day2000). Ne (with SMM) showed a slowly declining trend over time, with values of 45 661, 41 969 and 22 896 respectively (95% CI ranging from 9839–104 013; Table 2). Estimates based on the IAM model were several times lower (7057–10 513, 95% CI = 4716–25 000).
Recently bottlenecked populations are more likely to lose rare alleles leading to a mode-shift distortion in the distribution of allele frequencies at neutral loci such that alleles in the low frequency class (<0·1) become less abundant than alleles in one or more intermediate frequency classes (Luikart et al. Reference Luikart, Allendorf, Cornuet and Sherwin1998). Contrary to this expectation, the Sri Lankan P. vivax population showed no evidence of a mode-shift in the allele frequency distribution with approximately 90% being rare alleles.
The reduction in allelic diversity is faster than heterozygosity, thereby leading to an excess of heterozygosity in a recently reduced population. We considered the population as a whole as well as according to the time periods 2003–4, 2005 and 2006–7. There was no heterozygosity excess seen with either the Sign test or the Wilcoxon sign rank test, assuming a TPM model with 70% stepwise mutations (P>0·29 and P>0·59 respectively; Table 2).
A genetic bottleneck was only detected with M-ratio, at all 14 loci (critical value = 0·68). M-ratios were low for each locus (0·15–0·33, mean 0·26; Table 1), reflecting that most allelic states were unoccupied at all 14 loci. M-ratios calculated for each locus according to the years of sample collection too, revealed similar results (Table 1). A decline in M-ratios was observed in 10 out of the 14 loci tested between the time periods from 2003–4 to 2006–7.
DISCUSSION
Parasite diversity provides opportunity for better adaptation and survival thereby facilitating transmission. Knowledge of parasite population genetics would therefore be useful in designing and monitoring strategies for its elimination. There are now five reference genomes of P. vivax that are published (Carlton et al. Reference Carlton, Adams, Silva, Bidwell, Lorenzi, Caler, Crabtree, Angiuoli, Merino, Amedeo, Cheng, Coulson, Crabb, Del Portillo, Essien, Feldblyum, Fernandez-Becerra, Gilson, Gueye, Guo, Kang'a, Kooij, Korsinczky, Meyer, Nene, Paulsen, White, Ralph, Ren, Sargeant, Salzberg, Stoeckert, Sullivan, Yamamoto, Hoffman, Wortman, Gardner, Galinski, Barnwell and Fraser-Liggett2008; Neafsey et al. Reference Neafsey, Galinsky, Jiang, Young, Sykes, Saif, Gujja, Goldberg, Young, Zeng, Chapman, Dash, Anvikar, Sutton, Birren, Escalante, Barnwell and Carlton2012), and a very interesting finding from the analysis of these P. vivax genomes is that the species exhibits almost twice as much genetic diversity than P. falciparum (Neafsey et al. Reference Neafsey, Galinsky, Jiang, Young, Sykes, Saif, Gujja, Goldberg, Young, Zeng, Chapman, Dash, Anvikar, Sutton, Birren, Escalante, Barnwell and Carlton2012). Analysis of SNPs and microsatellite markers suggest a globally higher genetic diversity in P. vivax than P. falciparum, indicating a distinct history of global colonization, a more stable demographic history and a capacity for greater functional variation for P. vivax relative to P. falciparum (Neafsey et al. Reference Neafsey, Galinsky, Jiang, Young, Sykes, Saif, Gujja, Goldberg, Young, Zeng, Chapman, Dash, Anvikar, Sutton, Birren, Escalante, Barnwell and Carlton2012). The implications of this as regards to control and eventual elimination of vivax malaria are enormous, warranting deeper study of these variations of P. vivax so that intervention strategies can target its distinctive biology.
Due to implementation of various parasitological and entomological interventions, Sri Lanka has successfully reduced its malaria case load by 99·99% over the past decade (Anti-Malaria Campaign, Sri Lanka). A dramatic reduction in census population size is expected to result in increased inbreeding, diminished heterozygosity, increased geographical differentiation and reduced effective population size. Our P. vivax population did not conform to most of these expected changes. We observed high levels of heterozygosity and large effective population sizes over a period of time during which malaria transmission has drastically declined in Sri Lanka. Moreover, local parasite populations are characterized by high levels of allelic polymorphisms, a large proportion of multiple clone infections and significant LD indicative of reduced meiotic recombination, together with marked differentiation of parasites, both in time and space.
The level of genetic variation represents a balance between mutation, drift and natural selection. Genetic variation is generated by mutation and is lost by genetic drift. Natural selection may either erode genetic variation by leading to fixation of alleles or promote its retention as a result of balancing or diversifying selection (Frankham, Reference Frankham1996). In our population a genetic bottleneck was detected by only one test (M-ratio). Both the mode shift test and the bottleneck programme exploits the fact that rare alleles are lost first through genetic drift while the M-ratio test additionally considers the overall range of allele sizes as well. So these findings can be interpreted as the loss of some rare alleles without affecting the allele size range and rare alleles maintained as a significant proportion of our population. It is also possible that the 14 loci tested did not have enough power to detect recent or weak reductions in population size despite having exceeded the minimum of 10 recommended for Bottleneck tests. Furthermore, the programme Bottleneck has recently been shown to have a false-negative period spanning 2–4Ne generations, necessitating a gap of more than several thousand generations separating the event and sampling before a bottleneck could be reliably detected (Cristescu et al. Reference Cristescu, Sherwin, Handasyde, Cahill and Cooper2010).
Our estimates of the effective population size using the method of Anderson et al. (Reference Anderson, Haubold, Williams, Estrada-Franco, Richardson, Mollinedo, Bockarie, Mokili, Mharakurwa, French, Whitworth, Velez, Brockman, Nosten, Ferreira and Day2000) gave relatively high values, but appeared to be declining over time (2003–4 to 2006–7). Estimates of effective population size based on heterozygosity reflect long-term processes because changes in H E occur very slowly over time and therefore, may not be a reliable indicator for studying contemporary demographic processes. With the drop in incidence of malaria in Sri Lanka our sample numbers varied greatly both in time and space. When true Ne is large (>100), fluctuations in allele frequency may be very small, so that large samples sizes are required to measure Ne with accuracy (Nkhoma et al. Reference Nkhoma, Nair, Al-Saai, Ashley, McGready, Phyo, Nosten and Anderson2012). As such it is possible that insufficient sampling may have led to bias in the estimates of effective population size (Palstra and Ruzzante, Reference Palstra and Ruzzante2008). It is also possible that our analysis lacked sufficient power to detect a decline in Ne since changes in high Ne are more difficult to determine with precision, although it is improved by the use of diverse markers (Palstra and Ruzzante, Reference Palstra and Ruzzante2008). Power analyses of the temporal method for detecting population bottlenecks reveal that substantial numbers of loci and individuals (>10 loci, S>45) would enhance the validity of studies (Luikart et al. Reference Luikart, Cornuet and Allendorf1999) therefore the use of 14 highly diverse loci in our analyses together with samples that were pooled to improve on the numbers are likely to have strengthened the results.
Although recently bottlenecked populations are likely to have lost rare alleles, they may still contain substantial heterozygosity and quantitative genetic variation which are lost more slowly than allelic variation, and may influence fitness in current environments more than allelic variation (Luikart et al. Reference Luikart, Allendorf, Cornuet and Sherwin1998). High genetic diversity could be maintained due to several factors. Plasmodium vivax has a hypnozoite stage which lies dormant in the liver and can cause a relapse of the illness long after the primary episode has been cleared. The activation of heterologous hypnozoites can lead to increased genetic diversity (Imwong et al. Reference Imwong, Snounou, Pukrittayakamee, Tanomsing, Kim, Nandy, Guthmann, Nosten, Carlton, Looareesuwan, Nair, Sudimack, Day, Anderson and White2007b ). Population migration and tourism in Sri Lanka too could increase admixture of parasite strains and clones resulting in high genetic diversity. And asymptomatic parasite reservoirs, although shown to be significant in the past (Kodisinghe, Reference Kodisinghe1991; Gunawardena, Reference Gunawardena1998), have not been evident in more recent studies (Fernando et al. Reference Fernando, Abeyasinghe, Galappaththy and Rajapaksa2009; Rajakaruna et al. Reference Gunawardena, Karunaweera, Ferreira, Phone-Kyaw, Pollack, Alifrangis, Rajakaruna, Konradsen, Amerasinghe, Schousboe, Galappaththy, Abeyasinghe, Hartl and Wirth2010).
Haploid malaria parasites replicate mitotically in the human host, with some cells differentiating into male and female sexual stages, the gametocytes. The sexual phase takes place within the mosquito midgut, following ingestion of gametocytes by the vector mosquito during a blood meal. Male and female gametes fuse to form a short-lived diploid zygote, which then undergoes meiosis to generate haploid infective stages. Recombination occurs during the brief sexual stage resulting in the re-assortment of genes and generation of new parasite genotypes. Inbreeding or selfing occurs when male and female gametes of the same genotype fuse and the haploid infective stages that are generated are unchanged by recombination. When two genetically distinct gametes fuse, outbreeding occurs, and the resulting genome of the infective stage is reshuffled. In a declining population, inbreeding is increased and is characterized by: a loss of unique genotypes and an increase in identical genotypes; decline in MCI and the presence of significant LD. These features have been clearly demonstrated for declining populations of P. falciparum recently, in Senegal (Daniels et al. Reference Daniels, Chang, Sene, Park, Neafsey, Schaffner, Hamilton, Lukens, Van Tyne, Mboup, Sabeti, Ndiaye, Wirth, Hartl and Volkman2013) as well as on the Thai–Burma border (Nkhoma et al. Reference Nkhoma, Nair, Al-Saai, Ashley, McGready, Phyo, Nosten and Anderson2012). Contrastingly, our P. vivax population demonstrated almost all unique genotypes with high proportions of MCI accompanied by significant levels of LD.
Plasmodium vivax in low transmission areas has displayed both high genetic diversity and significant LD previously (Ferreira et al. Reference Ferreira, Karunaweera, Silva-Nunes, Da Silva, Wirth and Hartl2007; Imwong et al. Reference Imwong, Nair, Pukrittayakamee, Sudimack, Williams, Mayxay, Newton, Kim, Nandy, Osorio, Carlton, White, Day and Anderson2007a ; Karunaweera et al. Reference Karunaweera, Ferreira, Munasinghe, Barnwell, Collins, King, Kawamoto, Hartl and Wirth2008). Here too we observed strong LD being maintained both geographically and temporally (Tables 2 and 3) as well as between individual pairs of loci situated either in the same or in different chromosomes. Significant deviations from random association among loci suggest that the rate of effective recombination may be low relative to mutation, such that LD is maintained. Strand slippage events in microsatellites might occur so frequently during mitotic replication of parasites that new haplotypes may be generated without affecting the overall patterns of association between loci (Levinson and Gutman, Reference Levinson and Gutman1987; Karunaweera et al. Reference Karunaweera, Ferreira, Munasinghe, Barnwell, Collins, King, Kawamoto, Hartl and Wirth2008). High diversity and strong LD has been observed among Sri Lankan isolates using single-nucleotide polymorphism (SNP) analysis as well (Orjuela-Sanchez et al. Reference Orjuela-Sanchez, Karunaweera, da Silva-Nunes, da Silva, Scopel, Goncalves, Amaratunga, Sa, Socheat, Fairhurst, Gunawardena, Thavakodirasah, Galapaththy, Abeysinghe, Kawamoto, Wirth and Ferreira2010).
A high carriage of multiple clone infections was detected in our P. vivax population. Varying efficiencies of PCR amplification are known to cause biases that can lead to inaccurate assignment of predominant haplotypes (Havryliuk et al. Reference Havryliuk, Orjuela-Sanchez and Ferreira2008). Minor alleles may also be confused with artefacts such as stutter peaks or unspecified PCR amplification products (Havryliuk and Ferreira, Reference Havryliuk and Ferreira2009). To overcome this, a minimum value of at least one third of the predominant allele peak was used as a cut off for scoring minor alleles. The level of MCI was reduced greatly (Table 2) when multiple alleles at only two or more loci were considered. Extensive clonal diversity of P. vivax infection in areas with relatively low transmission has been observed previously as well (Ferreira et al. Reference Ferreira, Karunaweera, Silva-Nunes, Da Silva, Wirth and Hartl2007; Imwong et al. Reference Imwong, Snounou, Pukrittayakamee, Tanomsing, Kim, Nandy, Guthmann, Nosten, Carlton, Looareesuwan, Nair, Sudimack, Day, Anderson and White2007b ). Parasite relapses provide a biological basis for increased clonal diversity. Reactivation of hypnozoites while superinfected with a new strain can give rise to genetically distinct strains detectable in the bloodstream. And competition between co-infecting parasite clones with selection for favourable phenotypes may have significant implications for elimination strategies (Havryliuk and Ferreira, Reference Havryliuk and Ferreira2009).
We observed fragmentation of the population as evidenced by low but significant genetic differentiation (P⩽0·0009) both in time and space (Tables 4–6). Fragmentation reduces population size and increases isolation and the remnant populations experience increased genetic drift and inbreeding (Frankham et al. Reference Frankham, Ballou and Briscoe2003). Similar spatial and temporal substructuring in Sri Lankan parasites was also revealed by Schousboe et al. (Reference Schousboe, Rajakaruna, Amerasinghe, Konradsen, Ord, Pearce, Bygbjerg, Roper and Alifrangis2011) with the use of 3 markers (Pvmsp-3α, m1501 and m3502) on isolates from 9 districts.
When a population is subdivided the amount of its genetic relatedness will depend on the amount of gene flow that takes place among the subpopulations or subgroups. Gene flow can efficiently balance the forces of inbreeding and genetic drift and restore genetic variation over time (Hedrick, Reference Hedrick2004). Thus importation of malaria parasites can undermine the elimination efforts conducted so far in Sri Lanka. The conducive climate prevailing throughout the year resulting in an abundance of vectors, a large population with little or no immunity to malaria (due to low prevalence during the past decade), and a growing number of foreign arrivals in the country have increased the likelihood that imported malaria could possibly be the most likely source of a resurgence of disease transmission in the country (Galappaththy et al. Reference Galappaththy, Fernando and Abeyasinghe2013).
Sri Lanka has experienced an increase of 176% in cases of imported malaria between 2008 and 2012, with the majority of cases being Sri Lankan nationals who have acquired infection mainly from the South Asian region, especially India (Galappaththy et al. Reference Galappaththy, Fernando and Abeyasinghe2013). Malaria surveillance has been greatly strengthened since the onset of the pre-elimination phase in 2008. All cases of malaria have to be reported to the Anti Malaria Campaign headquarters (AMC), after which follow-up of patients is done by the AMC. Individual case reporting, as well as 24 h case reporting systems are in place (Abeyasinghe et al. Reference Abeyasinghe, Galappaththy, Smith Gueye, Kahn and Feachem2012). Health centres specializing in malaria diagnosis offer voluntary screening for persons with fever at ports of entry to the country, while travellers departing Sri Lanka to malaria-endemic countries are encouraged to obtain prophylaxis (issued free of charge) available in the health centre at the airport or from the AMC headquarters (Galappaththy et al. Reference Galappaththy, Fernando and Abeyasinghe2013). We emphasize the need for maintenance of these disease surveillance measures which facilitate early detection and radical cure of cases as well as educating the public regarding the importance of compliance, in order to prevent reintroduction of malaria through importation.
The implications of high diversity of this P. vivax population on the feasibility for malaria elimination efforts are considerable. At the outset, a sustained high diversity could simply imply that the extent to which malaria transmission declined may have been grossly overestimated, though the failure in detection and/or reporting of cases of malaria seems unlikely given the strong public health system existing in the country. However, it would be prudent to be mindful that the efficacy of screening is essentially limited by the level of sensitivity of methods used. On the other hand, imported malaria infections (characterized by divergent genotypes) may have been misclassified as indigenous, artificially inflating the estimates of local parasite diversity. Generally all reported cases are investigated by the AMC through their Regional Malaria Officers to determine whether transmission was local or imported and recorded appropriately. Alternatively, these results may indicate that malaria parasites remained highly diverse despite real decline in demographic population size. If so, we can expect that a much more drastic reduction in population size would be required to have a major impact on these parameters, which in turn provide the parasite with the means necessary to survive in a changing environment (the most diverse parasites are the most likely to survive drug treatment or acquired immunity leaving at least some extant strains), emphasizing the need for continued maintenance of control and surveillance measures to prevent resurgence.
In the Nkhoma et al. (Reference Nkhoma, Nair, Al-Saai, Ashley, McGready, Phyo, Nosten and Anderson2012) study, the proportion of multiple clone infections was the most sensitive indicator of declining malaria transmission. However, no major change was observed with this parameter over time in our study. Therefore, it appears that more samples (over decades) might be required to detect the genetic signatures of shrinking malaria transmission. In conclusion, importation of malaria cases from neighbouring countries might have offset gains in local transmission reduction, and insufficient numbers of samples may have led to imprecise estimation of some genetic parameters. Emphasis is therefore placed on the need for an active case-finding programme and the screening of incoming travellers for malaria.
ACKNOWLEDGEMENTS
We thank the Director and staff of the National Malaria Control Programme, Sri Lanka, for helping with sample collection. We also thank Ms Chamika de Silva and Ms Dilusha Fernando from the Department of Parasitology, University of Colombo for technical assistance with the statistical software, and Dan Neafsey of the Broad Institute, USA, for useful comments regarding the manuscript.
FINANCIAL SUPPORT
This study received financial support from the National Institutes of Health (5R03TW007966-02), USA, and the National Research Council, Sri Lanka (NRC grant no. 09-02).
SUPPLEMENTARY MATERIAL
To view supplementary material for this article, please visit http://dx.doi.org/S0031182013002278.