INTRODUCTION
Leishmaniasis, caused by the sandfly-transmitted intracellular protozoan parasite Leishmania, occurs worldwide. An estimated 12 million people are infected with a yearly incidence of 1–1·5 million cases of cutaneous leishmaniasis and 500000 cases of visceral leishmaniasis (Desjeux, 1996). If untreated, the visceral form principally caused by Leishmania donovani is usually fatal. Due to the lack of vaccines, difficulties in vector control, and the development of parasite resistance toward several treatments, leishmaniasis represents a significant clinical and public health problem (Herwaldt, 1999). Development of new anti-leishmania drug alternatives is required, but a better understanding of the resistance mechanisms is crucial.
Eukaryotic DNA topoisomerase I (Topo-I) is an essential enzyme that regulates the topological changes of DNA that accompany DNA replication, transcription, recombination, and chromosome segregation during mitosis (Gellert, 1981; Wang, 1985, 1991). Topo-I introduces transient single-stranded DNA breaks in one of the phosphodiester backbones of the duplex DNA and results in a reversible Topo-I/DNA covalent complex (Champoux, 1976, 1978, 1981). In 1999, we cloned and sequenced the first Topo-I gene from L. donovani (LdTOP1A) (Broccoli et al. 1999). More recently, a novel Topo-I gene has also been described in this parasite (LdTOP1B) (Villa et al. 2003). These latter observations suggested a dimeric Topo-I in Leishmania parasites since the enzymatic activity was only detected when both genes (LdTOP1A and LdTOP1B) were co-expressed in a yeast expression system (Villa et al. 2003). Interestingly, a recent study demonstrated, for the first time, the in vitro reconstitution of two recombinant proteins, LdTOP1L and LdTOP1S, corresponding to the large (LdTOP1A) and small (LdTOP1B) subunits, respectively (Brata Das et al. 2004). Moreover, this study also revealed the localization of the active enzyme (LdTOP1LS) in both the nucleus and kinetoplast of the parasite (Brata Das et al. 2004).
Several DNA minor groove–binding ligands (MGBLs) have been reported to exhibit antitumor activity and act as Topo-I inhibitors (Chen et al. 1993a,b). Indeed, compounds such as the bisbenzimidazoles Hoechst 33342 (Ho342) and Hoechst 33258 (Ho258), and various terbenzimidazole derivatives have been identified as specific Topo-I inhibitors (Chen et al. 1993a,b; Sun et al. 1994, 1995; Kim et al. 1996b; Rangarajan et al. 2000). The MGBLs exert their principal action by interfering with the catalytic cycle of this enzyme during the creation of Topo-I-linked DNA breaks, thus leading to arrest of replication (Chen et al. 1993b). It is the stabilization of the cleavable complex that is responsible for DNA fragmentation and cytotoxicity (Liu, 1989). Recently, we reported that Ho342 targets Leishmania Topo-I, leading to parasite growth inhibition in vitro by mechanisms involving DNA breakage and an apoptotic-like phenomenon (Marquis, Drolet & Olivier, 2003). We also provided preliminary evidence that the resistance to Ho342 in L. donovani (LdRHo.50) seems to be conferred by an increase in Topo-I gene expression reflected by a higher level of Topo-I DNA relaxation activity (Marquis et al. 2003). As the resistance to Topo-I inhibitors in mammalian cells is usually multifactorial and involves mechanisms associated with drug transport (Endicott & Ling, 1989), drug–target interaction (Beck, 1987) and drug detoxification (Deffie et al. 1988), we became interested in increasing the parasite (LdRHo.300) resistance level against this drug and deciphering the mechanism(s) by which the parasites bypassed the cytotoxic effect of the Ho342.
Previous studies have reported that Ho342 was among the most effective MGBL compounds to induce Topo-I-cleavable complexes (Chen et al. 1993b; Sun et al. 1994; Kim et al. 1996a). Knowing that topoisomerases play a pivotal role in protozoan parasite replication (Cheesman, 2000) and that the Ho342 lead molecule (2,5′-Bi-1H-benzimidazole) (Kim et al. 1996a) can be used as a starting structure for derivative compounds more effectively, defining the Ho342 resistance mechanism(s) in Leishmania represents an important strategic tool. In the present study, we report the first observations on Ho342 resistance mechanisms occurring in protozoan parasites. Our findings may help to improve our knowledge on the Ho342 action mechanism on Leishmania Topo-I and to gain insight into the development of new potential derivatives in leishmaniasis chemotherapy.
MATERIALS AND METHODS
Drug solutions
Ho342 was purchased from Sigma and stock solution (25 mM) in dimethyl sulfoxide (Me2SO; ICN Biomedicals, Inc.) was stored at −20 °C.
Parasite cultures
The parasites (Leishmania donovani strain 1S2D (strain 1S, clone 2D, WHO designation: MHOM/SD/62/1S-CL2D)) were grown at room temperature and transferred bi-weekly in SDM-79 culture medium (SDM) supplemented with 10% fetal bovine serum (FBS) as previously described (Olivier & Tanner, 1987; White et al. 1988). Resistant parasites were selected for Ho342 (LdRHo.300) resistance by stepwise increase until reaching 300 μM. LdRHo.300 parasites were developed from L. donovani strain 1S2D. As a control, 3 other Ho342 resistant strains were also developed from 3 different clones of L. donovani strain 1S2D. However, since each clone produced similar results, we have only presented the data originating from 1 parasite strain.
Topo-I treatment
To monitor the impact of Topo-I inhibitors on Leishmania growth, parasites were transferred (2×106 log phase promastigotes/ml) into 3 ml of SDM in the absence or presence of increasing concentrations (0–50 μM) of Ho342. The growth was monitored over 6 days by measuring the absorbance at 600 nm using an automated microplate reader (Organon Teknika, Reader 510) (White et al. 1988). The data presented are representative of 3 experiments carried out in triplicate.
DNA sequencing
Sequencing of the Topo-I genes was carried out using a cycle sequencing kit (Big Dye Terminator cycle sequencing ready reaction, Perkin Elmer, CA) and an automated DNA sequencer (ABI Prism 377 DNA Sequencer, Perkin Elmer, CA).
Topo-I gene expression
Total RNA from 5×108 log phase promastigotes was extracted using TRIzol reagent (GIBCO BRL). Briefly, RNA was resolved on a 1% agarose gel and transferred to a Nytran® Plus nylon membrane. After material transfer, the membrane was UV-exposed for 3 min on a transilluminator, and pre-hybridized for 4 h at 42 °C in a solution of 5× SSC, 10× Denhardt's solution, 50 mM NaPO4, 0·2 mM Dextran sulfate, 0·5% SDS, 133 mM glycine, 50% formamide with 150 μg/ml of salmon sperm DNA. The membrane hybridization was performed overnight at 42 °C with a [α-32P]-dCTP-labelled SacI DNA fragment from the L. donovani TOP1-like gene (LdTOP1A), as we previously described (Broccoli et al. 1999). After incubation, the membrane was washed 3 times with 2× SSC/0·1% SDS and 3 times with 0·1× SSC/0·1% SDS (15 min per wash, 42 °C). Autoradiography was performed using X-ray film (Kodak). The data presented are representative of 3 experiments independently performed.
Topo-I activity assays
Parasite extracts were obtained from 5×108 log phase promastigotes, and submitted to the Topo-I activity assays (Tosh et al. 1999; Marquis et al. 2003). Briefly, the parasites were resuspended in 100–200 μl of Topo-I assay buffer (10 mM Tris-HCl, pH 7·9, 1 mM EDTA, 150 mM NaCl, 0·1% BSA, 0·1 mM spermidine, 5% glycerol), and lysed by repeated passage through a 25-gauge needle. The parasite extracts (2 μg of total proteins) were incubated for 1 h at 37 °C in a 20 μl volume containing Topo-I assay buffer and 0·5 μg of supercoiled pBR322 (Roche). They were further incubated with 0·5 μg/μl Proteinase K, 1% SDS, and 100 mM EDTA for 30 min at 50 °C before the addition of 2·5 μl of stop mix (5% sarkosyl, 0·0025% bromophenol blue, 25% glycerol). Supercoiled and relaxed forms of pBR322 were separated in a 1% agarose slab. After migration in 1× TBE buffer (89 mM Tris base/89 mM boric acid/2 mM EDTA, pH 8·0), the gel was soaked in ethidium bromide, and UV illuminated to reveal the status of DNA coiling. To exclude the possibility of Topo-II activity, Mg2+ ions and ATP were omitted from the reaction mix, both of which are well recognized to be necessary for Topo-II activity (Liu, Liu & Alberts, 1979; Stetler, King & Huang, 1979). All assays were repeated 3 times.
Drug accumulation and cellular integrity
Log phase promastigote parasites (2×106 parasites/ml) treated with Ho342 (0–100 μM) were collected at different time-points over 1 h. The effect of energy deprivation on Ho342 transport was evaluated by pre-treating the parasites with 20 mM sodium azide (NaN3) and/or by pre-incubating the parasites at 4 °C for 30 min. The fluorescent property of the Ho342 enabled the parasite fluorescence intensity and the cellular integrity to be directly evaluated by flow cytometry with an Epics Elite ESP (Coulter Electronics, Miami, Florida). The data obtained are representative of 3 experiments independently performed in triplicate.
Membrane protein analysis
Mid-log phase promastigote parasites were disrupted by sonication in PBS containing a protease inhibitor cocktail (Roche). After centrifugation to remove unbroken cells (3000 g, 15 min), the supernatant fractions were then ultracentrifuged (120000 g, 1 h). The resulting pellets were homogenized with 5 M urea (plus complete), incubated on ice for 1 h, and ultracentrifuged (120000 g, 30 min). These pellets were then homogenized in lysis buffer (7 M urea, 2 M thiourea, 4% CHAPS, 20 mM DTT, and 0·5% Triton X-100) and incubated overnight at room temperature. Following ultracentrifugation, the final pellets were homogenized in urea cracking buffer (5 M urea, 10 mM NaH2PO4, 1% SDS, 1% beta-mercaptoethanol, pH 7·0). Protein concentrations were determined by Amido Black assay. Proteins were aliquoted into single-use samples and stored at −80 °C. Proteins (50 μg) were migrated on 12% acrylamide gel (SDS-PAGE) and visualized by either silver staining (Silver Staining Kit from Pharmacia Biotech) or Sypro Ruby fluorescence. For Sypro Ruby, gel was fixed for at least 2 h in 10% methanol, 7% acetic acid. Gel was then stained for 5 h to overnight and destained in fixing solution. Gel was scanned using a Typhoon laser scanner (Molecular Dynamics). Protein migration patterns are representative of 3 experiments independently performed.
Mass spectrometry
After Sypro Ruby staining, gel plugs containing the proteins of interest were excised and sent for mass spectrometry analysis (Eastern Quebec Proteomics Center, Centre Hospitalier de l'Université Laval, Québec, Canada). Tryptic digestions of gel plugs were performed on a MassPrep liquid handling robot (Micromass) according to the manufacturer's specifications using sequencing grade modified trypsin (Promega). Peptide tandem mass spectra were obtained by capillary liquid chromatography coupled to an LCQ DecaXP (ThermoFinnigan, San Jose, CA) quadrupole ion trap mass spectrometer with a nanospray interface. Resulting peptide MS/MS spectra were interpreted using the SEQUEST algorithm and searched against proteins in the National Center for Biotechnology Information (NCBI) non-redundant protein database. A protein was considered a good match if at least 2 peptides were confidently identified.
RESULTS
Sensitivity of wild-type and resistant parasites to Ho342
Drug resistance mechanisms in Leishmania are usually investigated by analysing mutants selected for resistance by increasing drug concentrations (Borst & Ouellette, 1995). In order to decipher the resistance mechanisms occurring in Ho342 resistance, we have selected parasites for resistance to this drug in a step-by-step manner until they reached a resistance level of 30-fold over their wild-type counterparts. To compare their susceptibility to Ho342, we exposed freshly harvested parasites to increasing concentrations of the drug. Whereas the LdRHo.300 survival was not affected by the cytotoxic effect of Ho342, the growth of the wild-type parasites was inhibited in a dose-dependent manner with an ED50 of approximately 10 μM (Fig. 1). This difference of phenotype clearly established the development of resistance mechanisms in the resistant strain to circumvent the cytotoxic effect of this compound.
Fig. 1. Effect of Ho342 on Leishmania donovani growth. Ld 1S2D and LdRHo.300 were grown in the presence of increasing concentrations (0, 10, 25 and 50 μM) of Ho342. Optical density was followed over 6 days (% over control +/−S.E.).
Topo-I gene sequencing
The resistance to Topo-I inhibitors in cancer cells is commonly associated with Topo-I gene mutations (Pommier et al. 1994). It is possible that during drug selection we may have selected for a point mutation within the Topo-I genes (LdTOP1A and LdTOP1B) which is responsible for the observed resistance phenotype. To address this possibility, we proceeded to the Topo-I genes (LdTOP1A and LdTOP1B) sequencing of the resistant parasites, and compared it to the sequence of the genes derived from susceptible parasite strain. We concluded that no mutation was observed in the LdTOP1A and LdTOP1B genes of LdRHo.300 (data not shown).
Topo-I gene expression and enzymatic activity
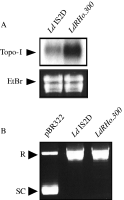
Since we recently correlated that the decrease of resistant parasite (LdRHo.50) susceptibility to Ho342 was potentially attributed to an increase of the Topo-I gene (LdTOP1A) expression (Marquis et al. 2003), we thought it was important to further corroborate this observation with the LdRHo.300 strain. The amount of LdTOP1A mRNA in the wild-type and resistant parasites was determined by Northern blot analysis. As reported in Fig. 2A, a 3·4 kb LdTOP1A mRNA present in Ld 1S2D was around 3-fold more expressed in LdRHo.300.
Fig. 2. Topo-I gene (LdTOP1A) expression and Topo-I DNA relaxation activity in LdRHo.300: (A) Northern blot of Ld 1S2D and LdRHo.300. The probe used corresponds to the SacI DNA fragment (1069 bp) from the TOP1-like gene (LdTOP1A) of Leishmania donovani. The LdTOP1A mRNA is 3·4 Kb long according to molecular weight markers. RNA integrity was verified by ethidium bromide (EtBr) staining. (B) Topo-I DNA relaxation activity assay. Supercoiled pBR322 DNA alone is shown in lane 1. Supercoiled pBR322 DNA in the presence of parasite extracts are represented in the following lanes. R (Relaxed DNA form), SC (Supercoiled DNA form).
In the light of this result, we were interested to evaluate the impact of the Topo-I gene overexpression in LdRHo.300 on its Topo-I DNA relaxation activity. Thus, we measured the Topo-I activity contained in the wild-type and resistant parasites by the ATP-independent relaxation of supercoiled pBR322 DNA. As demonstrated in Fig. 2B, L. donovani 1S2D and LdRHo.300 were both capable to maximally relax the supercoiled DNA. In addition, we also performed serial limiting dilutions of each extracts for the same amount of protein, to determine which dilutions were able to abrogate the relaxation activity present in the extracts. As reported in Fig. 3A, whereas a dilution of 1[ratio ]4 was needed to restore the supercoiling level in the wild-type parasite, a 1[ratio ]16 dilution was necessary for LdRHo.300, suggesting a higher Topo-I activity in the latter. To further support this observation, we achieved Ho342 treatments with increasing concentrations of the drug on equal amounts of parasite extract. From this, we were able to observe that a dose of 20–25 μM was required to start the inhibition of DNA relaxation in LdRHo.300 extract, while only 5 μM Ho342 was necessary in the wild-type extract (Fig. 3B).
Fig. 3. Topo-I DNA relaxation activity of Ld 1S2D and LdRHo.300. (A) Effect of serial extract dilutions on Topo-I activity. Supercoiled pBR322 DNA alone is shown in lane 1. Supercoiled pBR322 DNA in the presence of parasite extract dilutions are represented in the following lanes (Ld 1S2D, lanes 2–7; LdRho.300, lanes 8–13). (B) Effect of Ho342 on Topo-I activity. Supercoiled pBR322 DNA alone is shown in lane 1. Supercoiled pBR322 DNA in the presence of parasite extracts and increasing concentrations of Ho342 (0–50 μM) are represented in the following lanes. R (Relaxed DNA form), SC (Supercoiled DNA form).
Altogether, these results support that there was more enzyme in the resistant parasite than in the wild-type, and consequently more Topo-I activity. Furthermore, this observation corroborates the fact that there was a higher Topo-I gene expression in LdRHo.300. As the resistance to Topo-I inhibitors is usually multifactorial, in the following sections, we thus performed experiments to establish whether or not the resistance to Ho342 could involve other resistance mechanisms. The fact that resistant parasites were selected for in a stepwise fashion could also have contributed to a multifactorial response.
Ho342 accumulation alteration in LdRHo.300
One possible way to increase the level of resistance would be by blocking the drug uptake. To test this possibility, we directly measured the Ho342 accumulation inside the parasites and, in parallel, we evaluated the cellular integrity during the course of treatments. Flow cytometry analysis was performed to determine whether or not the wild-type strain was more permissive to the Ho342 accumulation than the resistant parasites. While L. donovani 1S2D accumulated the Ho342 in a dose-dependent manner, there was practically no accumulation of the drug inside LdRHo.300 (Fig. 4A). Indeed, at 25, 50, and 100 μM the fluorescence intensity of LdRHo.300 was blocked at the same level, clearly showing a limited accumulation of the drug inside the parasites. If we focus on the 100 μM-Ho342 treatments, we can observe that there was 5 to 6-fold less drug accumulation inside LdRHo.300 than L. donovani 1S2D. Additionally, the Ho342 accumulation inside the parasites was directly proportional to the decrease of parasite viability (Fig. 4B). To deepen our investigation on this difference of accumulation, we evaluated the effect of energy deprivation on 100 μM-Ho342 transport by incubating the parasites at 4 °C and/or by treating the parasites with the metabolic inhibitor sodium azide (20 mM NaN3) (Kundig et al. 1999). Similar concentration of NaN3 was shown to inhibit active folate uptake in Leishmania (Ellenberger & Beverley, 1987). While NaN3 was capable of reducing the Ho342 accumulation inside the wild-type strain by approximately 50%, the incubation at 4 °C decreased the accumulation at the same level than for LdRHo.300 (Fig. 5A). Moreover, the wild-type parasite treatment with NaN3 in combination with 4 °C incubation had no significant effect on Ho342 accumulation over incubation at 4 °C alone (Fig. 5A). Interestingly, we observed that this decrease of drug accumulation conferred by NaN3 treatment or by the incubation at 4 °C was able to proportionally protect the wild-type parasite against the cytotoxic effect of the Ho342 (Fig. 5B). However, as there was practically no Ho342 accumulation in the resistant parasites, NaN3 treatment and the incubation at 4 °C had no significant effect on drug accumulation (Fig. 5A). Together, all these results suggest the presence of a potential energy-dependent Ho342 membrane transporter in the wild-type strain, and that a modification of this transporter has occurred in the resistant parasite, leading to an altered drug accumulation.
Fig. 4. Ho342 permeability and parasite viability in LdRHo.300. (A) Ho342 permeability. Ld 1S2D and LdRHo.300 treated with Ho342 (0–100 μM) were collected at different time-points over 1 h and analysed by flow cytometry. The fluorescence intensity (A) results are expressed in arbitrary units (mean +/−S.E.), and viability (B) in percentages.
Fig. 5. Effect of NaN3 and 4 °C incubation on Ho342 accumulation in LdRHo.300. Ld 1S2D and LdRHo.300 were treated with 100 μM of Ho342, in presence or not of 20 mM sodium azide (NaN3) and/or 4 °C incubation. The samples were collected at different time-points over 1 h and analysed by flow cytometry. The fluorescence intensity (A) results are expressed in arbitrary units (mean +/−S.E.), and viability (B) in percentages.
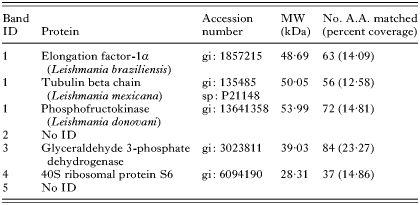
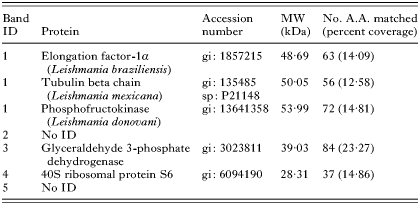
To further understand the potential mechanism underlying this drug accumulation alteration, we investigated the Ho342 transport system. By using a one-dimension gel approach, membrane protein profiles were compared between wild-type and Ho342 resistant parasites to determine whether change in membrane composition could be detected between those parasite strains. Indeed, this result revealed that at least 5 bands of proteins present in L. donovani 1S2D were substantially reduced in LdRHo.300 (Fig. 6). Following mass spectrometry (MS/MS) analysis, 3 of these 5 bands were identified (Table 1). A 32 kDa band revealed the identity of the 40S ribosomal protein S6. The glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was identified in a second band at 35 kDa. Finally, another band at 50 kDa contained 3 proteins corresponding to the elongation factor 1-alpha (EF-1α), the tubulin-beta chain (Leishmania mexicana), and the phosphofructokinase (L. donovani).
Fig. 6. Analysis of LdRHo.300 membrane proteins. Membrane proteins of Ld 1S2D and LdRHo.300 were extracted and compared on SDS-PAGE as described in the Materials and Methods section. After pre-selection on a silver-stained gel, 5 down-regulated protein bands (identified by the black arrows) were isolated from a Sypro Ruby stained gel and analysed by mass spectrometry (MS/MS).
DISCUSSION
In the present study, we provided experimental evidence for the characterization of L. donovani resistance mechanisms to Ho342; using LdRHo.300 parasites around 30-fold more resistant to Ho342 when compared to their wild-type counterparts. To study the mechanisms responsible for the development of resistance in LdRHo.300, a series of quantitative and qualitative assays on Topo-I were performed. Since the resistance to Topo-I inhibitors in cancer cells is commonly associated with Topo-I gene mutations (Pommier et al. 1994), we proceeded to the Topo-I genes (LdTOP1A and LdTOP1B) sequencing of LdRHo.300. However, LdRHo.300 did not show any Topo-I gene mutation when compared with the wild-type sequences.
On the other hand, Northern blot analysis showed an increase of Topo-I gene (LdTOP1A) expression in LdRHo.300, corroborating our result previously published (Marquis et al. 2003). The increase of Topo-I gene expression in LdRHo.300, was reflected by the Topo-I DNA relaxation activity of these resistant parasites. In conformity with our previous work with LdRHo.50 (Marquis et al. 2003), our experimental approach designed to evaluate the Topo-I activity in LdRHo.300 showed a higher level of enzyme activity compared to its wild-type counterpart. This increased enzymatic activity correlated with the Topo-I gene overexpression, resulting in a greater Topo-I enzyme production in LdRHo.300.
As the resistant parasites were selected for in a stepwise fashion and that resistance to topoisomerase inhibitors is usually multifactorial, we further investigated the presence of other potential resistance mechanisms. Because the Leishmania plasma membrane represents the first cellular barrier that Ho342 encounter before reaching its specific target, we determined the efficiency of the drug to accumulate inside the wild-type and resistant parasites, in order to verify differences in their Ho342 accumulation. Our results on Ho342 cellular accumulation allowed us to suggest the presence of a potential energy-dependent Ho342 transporter in the wild-type parasite, and that an alteration of this transporter has occurred in LdRHo.300, leading to an altered drug accumulation. As the Ho342 transport across the cellular membrane in mammalian cells is usually mediated by passive diffusion (Chen et al. 1993a), the observation of an active Ho342 transport in Leishmania parasites could suggest a novel transport mechanism for this compound.
The membrane protein analysis of LdRHo.300 compared to its wild-type counterpart allowed us to obtain the identification of 5 proteins (40S ribosomal protein S6, GAPDH, EF-1α, tubulin beta chain, and phosphofructokinase) by mass spectrometry (MS/MS). Despite the fact that none of these proteins were integrally part of the parasite membrane, it is not excluded that they can have interacted with membrane proteins and played a role in drug accumulation within the parasites. Nevertheless, these proteins seem to be closely related and may have a role to play in the Ho342 resistance. For example, microtubules (built from heterodimers of α- and β-tubulin monomers) are essential for a wide variety of cellular functions, notably in membrane transport. Tubulin has also been recognized to play a role in resistance to arsenite in L. donovani (Prasad & Dey, 2000; Jayanarayan & Dey, 2002), and to taxol in mammalian cells (Verdier-Pinard et al. 2003). Moreover, co-immunoprecipitation analysis in the ciliate Tetrahymena pyriformis has permitted to make an association between beta-tubulin and EF-1α (Nakazawa et al. 1999), which play a role in apoptosis (Billaut-Mulot et al. 1996; Duttaroy et al. 1998; Talapatra, Wagner & Thompson, 2002). Since we previously observed an apoptotic-like phenomenon occurring in Ho342-treated L. donovani (Marquis et al. 2003), it is consistent that EF-1α could be involved in the Ho342 resistance development. Furthermore, the GAPDH (Sirover, 1999) and the phosphofructokinase (Louassini et al. 1999; Wegener & Krause, 2002) were both recognized as key regulatory enzymes playing an important role in the limiting step of glucose catabolism which is crucial for energetic processes. In addition to its glycolytic function, GAPDH displays a number of diverse activities including a role in DNA replication and DNA repair (Sirover, 1999), explaining another possible role in Ho342 resistance development. In the light of these observations, the involvement of these proteins in glycolysis, in DNA replication and repair, in drug resistance and in cell death, suggests their potential participation to the Ho342 resistance in LdRHo.300. However, their contribution in the resistance still remains to be validated. Moreover, the use of a 2-dimension gel electrophoresis might give a better separation and resolution of the membrane protein and, in consequence, might identify more different proteins that could be involved in Ho342 resistance.
Taken together, our findings suggested that Ho342 resistance in L. donovani potentially involves complex resistance mechanisms, most likely dominated by a reduced drug accumulation. This is the first report concerning resistance mechanisms against Ho342 in protozoan parasites. Finally, these observations provide new insight into the general Ho342 resistance mechanism development.
We would like to thank Dr Marc Ouellette (Centre de Recherche en Infectiologie, Université Laval, Québec, Canada) for his participation in several research discussions, as well as Vicky Brochu (research assistant in the laboratory of Dr Ouellette) who kindly provided us the protocol of membrane protein isolation. M.O. is supported by funds from the Canadian Institute of Health Research (CIHR) and is a member of a CIHR group in host-pathogen interactions. M.O. holds a CIHR investigator award and is a Burroughs Wellcome Fund awardee in molecular parasitology. J.-F.M. and I.H. are the recipients of CIHR doctoral studentships.