



Introduction
The study of symbiosis, and particularly mutualistic endosymbiosis (defined as one organism living inside another in a cooperative arrangement) has been gaining importance over time as such relationships grow in number and diversity. While endosymbiosis has been long considered a central phenomenon in the evolution of eukaryotic life and the origin or organelles (for a review, see Archibald, Reference Archibald2015), it is also implicated in more recent, but still significant, biological interactions in organisms ranging from bacteria to single-celled eukaryotes to Metazoa (usually insects) and plants.
Endosymbionts frequently specialize in complementing the metabolic capabilities of the host (Douglas, Reference Douglas2016). For example, in the nested endosymbiosis involving two bacteria (Gammaproteobacteria within a Betaproteobacteria) within mealybug bacteriocytes (von Dohlen et al. Reference von Dohlen2001), each bacterium has retained just the genes necessary for supplying the insect with essential amino acids (McCutcheon and von Dohlen, Reference McCutcheon and von Dohlen2011), complementing each other as well as the insect host. Secondary endosymbiosis between eukaryotes has been responsible for the origin of complex organisms such as parasites of the phylum Apicomplexa, which usually contain a highly reduced algal cell (apicoplast) in the cytoplasm (Gentil et al. Reference Gentil2017). The study of endosymbiotic relationships is thus of great interest in the characterization of genetic, metabolic, ecological and evolutionary aspects.
The parasitic flagellates of the subfamily Strigomonadinae (Kinetoplastea: Trypanosomatidae) are gut-dwelling insect parasites bearing endosymbiotic bacteria from the family Alcaligenaceae (Betaproteobacteria) in their cytoplasm (Teixeira et al. Reference Teixeira2011; Votýpka et al. Reference Votýpka2014). Owing to this peculiarity, the Strigomonadinae are relatively well studied, although not to such extent as their relatives from the genera Trypanosoma and Leishmania, many of which are of medical or veterinary importance (Nussbaum et al. Reference Nussbaum2010).
As uncovered in studies of species from the genera Angomonas and Strigomonas, the collaboration between the endosymbiont and its trypanosomatid host is very close and finely tuned. There is typically only one bacterium inside each trypanosomatid cell, and the division of the two partners is synchronized (Motta et al. Reference Motta2010). Significantly, as shown by biochemical and cell biology experiments over decades and recently characterized at the genome level, the bacterium provides the eukaryotic host with several important nutrients, such as essential amino acids (Alves et al. Reference Alves2013b), vitamins (Klein et al. Reference Klein2013) and the haem group (Kořený et al. Reference Kořený, Lukes and Oborník2010; Alves et al. Reference Alves2011). The bacterium cannot survive outside its host, while the aposymbiotic (i.e. ‘cured’ by antibiotic treatment) trypanosomatid can grow only in a medium supplemented with those compounds previously provided by the endosymbiont.
Although these parasites have been studied for a long time, their taxonomic status has only recently been clarified through the use of molecular data (Teixeira et al. Reference Teixeira2011), placing them into the sister genera Angomonas and Strigomonas. Each of two Angomonas (A. deanei and A. desouzai) and three Strigomonas species (S. culicis, S. galati and S. oncopelti) carries its own species of Candidatus Kinetoplastibacterium (Ca. K. crithidii, Ca. K. desouzaii, Ca. K. blastocrithidii, Ca. K. galatii and Ca. K. oncopeltii, respectively). The sole exception is A. ambiguus, which bears the same species of endosymbiont as A. deanei, in spite of its nuclear genome being more closely related to A. desouzai. Apart from that ambiguity, the phylogenetic tree of these trypanosomatids shows an identical branching pattern to that seen for the corresponding symbionts, providing compelling evidence that there was a single origin for this endosymbiosis, followed by co-speciation (Teixeira et al. Reference Teixeira2011).
More recently, a third genus in this subfamily was discovered, containing so far only one described species: Kentomonas sorsogonicus (Votýpka et al. Reference Votýpka2014). This trypanosomatid, like all other Strigomonadinae, contains an intracytoplasmic endosymbiont, Ca. K. sorsogonicusi. Preliminary phylogenetic analyses of the host and the bacterium using their SSU rRNA genes showed conflicting results, with Kentomonas being sister to the rest of Strigomonadinae, but its symbiont clustering with those from Angomonas, thereby making the bacteria of Strigomonas spp. the earliest branch of the genus Ca. Kinetoplastibacterium. However, the relationships between endosymbionts were poorly supported (Votýpka et al. Reference Votýpka2014).
Herein, we characterize the complete genome of Ca. K. sorsogonicusi, showing that it is even more reduced in size than the genomes of other Ca. Kinetoplastibacterium spp., having lost the haem-synthesis pathway until now regarded as a hallmark of endosymbionts in trypanosomatids. We also resolve the phylogenetic position of this organism within its genus by phylogenomic methods.
Materials and methods
Organism cultivation and DNA isolation
For genomic DNA preparation, K. sorsogonicus strain MF08 was grown in the BHI medium as previously described (Votýpka et al. Reference Votýpka2014). Log-phase trypanosomatid cells were collected by centrifugation for 10 min at 1000 g, washed once with PBS and frozen at −20 °C until DNA isolation. The latter was performed using DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions.
Haem requirement tests were performed in the M199 medium (Life Technologies, Carlsbad, USA) without fetal bovine serum and supplemented with biopterin (2 µg mL−1), HEPES (25 mm) as well as a mix of microelements as described previously (Porcel et al. Reference Porcel2014). Log-phase PBS-washed trypanosomatid cells were seeded into flat-sided tubes with 2 mL of this medium both with and without 10 µg mL−1 of haemin (M199+ and M199−, respectively). After 3 days, 100 µL of the culture from the M199− tube were placed into new tubes with M199+ and M199−.
Genome sequencing and assembly
Shotgun genome sequencing of K. sorsogonicus and its symbiont was performed jointly, without separating the two organisms. Genomic sequencing (Macrogen Inc., Seoul, South Korea) was performed using an Illumina paired-end library (2 × 100, inserts of on average 600 bp) constructed using the TruSeq DNA PCR Free kit.
Reads were trimmed using cutadapt (Martin, Reference Martin2011) to remove adapters and low-quality regions, and assembled using the Newbler v.2.9 assembler (distributed by 454 Roche).
Contigs and scaffolds corresponding to the endosymbiont were initially separated from the host sequences by BLASTN (Camacho et al. Reference Camacho2009) sequence similarity searches with other, fully sequenced Ca. Kinetoplastibacterium genomes. A minimum of 80% sequence similarity along most of the contig was used as the threshold for considering it as part of the endosymbiont's genome.
The final order of the contigs on the bacterial genome was identified by PCR-amplification using PCRBIO Taq Mix Red (PCR Biosystems Ltd., London, UK) and custom primers annealing at the ends of contigs: Ks1F (5′-gttctcttatgatacctccagt-3′), Ks1R (5′-ctccctcctactataattgct-3′), Ks2F (5′-agcacattgtaatccaggta-3′), Ks2R (5′-ggattgctagtatggtagaagg-3′), Ks3F (5′-cacaatcaggtgtagcatgt-3′), Ks3R (5′-tcaaatactcatacacgtgc-3′), Ks4F (5′-acccatagtagcaaaaccag-3′), Ks4R (5′-cgacgttcaagaccagatac-3′), Ks56F (5′-agctaaccgatactaattgc-3′) Ks56R (5′-cacgttccgatatattactcac-3′). All generated fragments were sequenced directly by the Sanger method (Macrogen Europe, Amsterdam, The Netherlands) using the amplification primers as well as those annealing to 16S and 23S rRNA genes (see Votýpka et al. Reference Votýpka2014).
Genomic annotation and analysis
The finished genome was annotated automatically by the Prokka pipeline v. 1.12 (Seemann, Reference Seemann2014) and annotations were sampled for manual validation against previously annotated Ca. Kinetoplastibacterium genomes whose annotations had been manually curated (see below).
Pseudogenes and missing predictions were identified by BLASTX of intergenic regions against the NCBI non-redundant database, followed by a manual examination.
The assembled genomic sequence and its annotation are available under accession number CP025628 (NCBI BioProject PRJNA414463). Other genomic sequences used in this work are: Ca. K. blastocrithidii TCC012E (accession number CP003733.1), Ca. K. crithidii TCC036E (CP003804.1), Ca. K. desouzaii TCC079E (CP003803.1), Ca. K. galatii TCC219 (CP003806.1), Ca. K. oncopeltii TCC290E (CP003805.1) (all five from Alves et al. Reference Alves2013a), Achromobacter arsenitoxydans SY8 (AGUF00000000.1) (Li et al. Reference Li2012) and Taylorella equigenitalis MCE9 (CP002456.1) (Hebert et al. Reference Hebert2011).
Overall structural comparison of Ca. K. sorsogonicusi and selected Ca. Kinetoplastibacterium spp. was performed by genome alignment in MAUVE v. 2015-02-13 (Darling et al. Reference Darling, Mau and Perna2010). Gene-level comparisons were based on the orthology inference results of OrthoMCL (Li et al. Reference Li, Stoeckert and Roos2003) analyses. Comparative metabolic annotation analysis of the endosymbionts from A. desouzaii, K. sorsogonicus, S. culicis and S. oncopelti was performed with ASGARD (Alves and Buck, Reference Alves and Buck2007), using the KEGG (Ogata et al. Reference Ogata1999) and UniRef100 (Suzek et al. Reference Suzek2007) databases as references, followed by manual curation of results of interest. Circos (Krzywinski et al. Reference Krzywinski2009) was used for generating genome comparison plots.
Phylogenetic analysis
All predicted proteins from either just the Ca. Kinetoplastibacterium spp. or all eight genomes mentioned above were analysed by OrthoMCL (Li et al. Reference Li, Stoeckert and Roos2003) in order to find all single-copy genes that were present in all organisms considered in each analysis. All such orthologous groups (OGs) identified were then aligned by MUSCLE v. 3.8.31 (Edgar, Reference Edgar2004), with subsequent removal of ambiguously aligned positions by Gblocks v. 0.91b (Castresana, Reference Castresana2000). The resulting filtered alignments were concatenated into a supermatrix with FASconCAT-G v.1.04 (Kück and Meusemann, Reference Kück and Meusemann2010), after conversion of alignment files to the requirements of that program using in-house Perl scripts.
Partitioned phylogenetic analysis of the resulting supermatrix was performed by maximum likelihood using RAxML v. 8.2.11 (Stamatakis, Reference Stamatakis2014) and running 100 bootstrap pseudoreplicates. Protein substitution models were selected automatically by the program, estimated separately for each partition using a maximum-likelihood criterion. Additionally, partitioned Bayesian inference was performed with MrBayes v. 3.2.6 (Ronquist et al. Reference Ronquist2012) using two runs of four chains (three heated) each for 100 000 generations (while monitoring run convergence parameters), using the ‘mixed’ amino acid substitution model and discarding 25% of the generations as burn-in. Substitution rate heterogeneity for each partition was modelled with gamma-distributed rates for both maximum likelihood and Bayesian inference methods.
Phylogenetic trees were drawn with MEGA v. 7 (Kumar et al. Reference Kumar, Stecher and Tamura2016) and cosmetic adjustments were performed in the Inkscape vector editor (https://inkscape.org).
Results
Genomic characterization
Genomic sequencing by Illumina paired-end technology yielded about 22.8 million pairs of 100 bp reads, for a total of 4.57 billion base pairs (around 114-fold coverage of a 40 million base pair genome). The assembly resulted in seven contigs that could be identified, by similarity to other Ca. Kinetoplastibacterium genomes, as belonging to the endosymbiont. These sequences were used for the design of PCR primers to amplify the adjacent genomic regions. The obtained fragments were sequenced, leading to the generation of an unambiguously complete genome assembly.
The genome of Ca. K. sorsogonicusi contains 741 697 base pairs and an overall GC content of 25.22%. Coding regions have similar composition, at 25.49% GC on average, while ribosomal and transfer RNA genes present the much higher average GC content of 50.36% (Table 1). The overall genome organization of Ca. K. sorsogonicusi can be seen in Fig. 1. The total number of genes predicted to be present in the genome is 722, with 670 protein-coding genes, three pseudogenes, 39 genes for tRNAs, nine genes for rRNAs (distributed in three clusters), and the gene for the ssrA transfer-messenger RNA.

Fig. 1. Schematic representation of the structure of, and comparison between, the genomes of Ca. K. sorsogonicusi, Ca. K. desouzaii and Ca. K. oncopeltii. Starting from the outside, the different rings mean, in order: genomic coordinate representations; predicted genes (in grey, protein-coding genes; in red, rRNA genes; in blue, tRNA genes; in green, the ssrA genes; and in black, pseudogenes); highlights of the position of the genes participating in haem synthesis in each organism (in red, the gene for glutamyl-tRNA synthetase, which is not specific to haem synthesis; in blue, all others); GC skew plots (negative skew in red and positive skew in yellow). The central area of the schema shows lines connecting the different orthologous groups found in both Ca. K. sorsogonicusi and Ca. K. desouzaii (blue lines) or Ca. K. oncopeltii (orange lines); black lines highlight the linkage between haem-synthesis genes present in all three organisms.
Table 1. Overall genome and gene composition statistics

Genome codes: CKsor: Candidatus Kinetoplastibacterium sorsogonicusi; CKcri: Ca. K. crithidii; CKdes: Ca. K. desouzaii; CKbla: Ca. K. blastocrithidii; CKgal: Ca. K. galatii; CKonc: Ca. K. oncopeltii; Tequi: Taylorella equigenitalis MCE9.
a Percentage of the total genome length that is in genes.
b Numbers include only apparently intact coding sequences, excluding pseudogenes.
The ribosomal gene clusters are not identical, with only one of them (the last one, starting at around position 590 000) containing two tRNAs between the large subunit rRNA (LSU) and the small subunit rRNA (SSU) genes. The SSU, LSU and 5S rRNA genes are, in this order, present in each of the three ribosomal gene clusters.
At least one tRNA gene has been identified for each of the 20 amino acids present in natural proteins. Eleven of the amino acids can be ligated to only one predicted tRNA. Leucine is the amino acid with the most tRNA genes predicted, with five, while arginine and serine have four tRNA genes each.
OG inference of the six Ca. Kinetoplastibacterium spp. genomes yielded 769 OGs, of which 132 were absent from Ca. K. sorsogonicusi. Among these 132 OGs, 68 (Supplementary File 1) were found in all other five Ca. Kinetoplastibacterium spp. genomes analysed here.
Figure 1 shows that the overall gene distribution in the Ca. K. sorsogonicusi genome is typical of those of Ca. Kinetoplastibacterium spp., with genes tightly spaced. Its comparison with the genomes of Ca. K. desouzaii and Ca. K. oncopeltii (the longest and shortest ones, respectively, among the previously sequenced species of the genus) demonstrates their overall synteny, which can be also extrapolated to all other Ca. Kinetoplastibacterium genomes studied so far. The GC skew plots for the three genomes are similar to those previously seen for Ca. K. blastocrithidii and Ca. K. crithidii (Alves et al. Reference Alves2013a). A little more than half of each genome shows predominantly positive GC skew whilst the remainder exhibits mostly negative skew (Fig. 1).
The only protein-coding gene duplication present in the Ca. Kinetoplastibacterium spp. genomes involves the EF–Tu gene. It is shown in Fig. 1 by lines running outside the genome coordinate circle and connecting the gene copies, close to the end of each sequence. This duplication is not present in Ca. K. sorsogonicusi, in a situation similar to that seen in Ca. K. galatii, where one of the copies was pseudogenized. In Ca. K. sorsogonicusi, however, even the pseudogene is absent.
Phylogenetic analysis
The inference of OGs by OrthoMCL yielded 506 single-copy genes present in the six Ca. Kinetoplastibacterium spp. and two species from other genera of the family Alcaligenaceae. Individual OG alignments were concatenated into a single data matrix comprising 186 276 columns. After removal of ambiguously aligned positions, 159 196 columns (86% of the original amount) remained in the alignment.
Phylogenomic analysis of all eight organisms by maximum likelihood resulted in the tree presented in Fig. 2. The Bayesian inference consensus tree is identical in topology and has nearly identical branch lengths (not shown). The outgroups (A. arsenitoxydans and T. equigenitalis) were used for rooting the tree, with the root being placed on the branch leading to A. arsenitoxydans in accordance with the previous phylogenomic inference of the Betaproteobacteria (Alves et al. Reference Alves2013a). All bootstrap support values and posterior probabilities in the tree reached the maximum values of 100 and 1.0, respectively.

Fig. 2. Maximum-likelihood protein supermatrix phylogeny of 506 orthologous groups from Ca. Kinetoplastibacterium spp. and two other Alcaligenaceae bacteria as outgroups. Semi-colon-separated numbers on each node represent the maximum-likelihood bootstrap support value (up to 100) and the Bayesian inference's posterior probability (up to 1) for that node.
The three endosymbionts from Strigomonas spp. formed a single clade, while those from Angomonas composed another one. Ca. K. sorsogonicusi was placed as the earliest group to split from the rest of Ca. Kinetoplastibacterium spp. (Fig. 2).
Genomic analyses of metabolic pathways
Comparison of metabolic pathways revealed only one of them was significantly disrupted in Ca. K. sorsogonicusi, whereas most other differences in enzyme presence or absence were scattered across the metabolic network. The haem pathway contains only two out of nine expected specific enzymes: oxygen-independent coproporphyrinogen-III oxidase (hemN) and ferrochelatase (hemH), respectively the eighth and the tenth (and last) enzymes of the pathway (Fig. 3 and Supplementary File 2, map 00 860, Porphyrin and chlorophyll metabolism). The first enzyme shown in the pathway, glutamyl-tRNA synthetase (gltX), not specific for the haem-synthesis pathway, is also present in the genome.
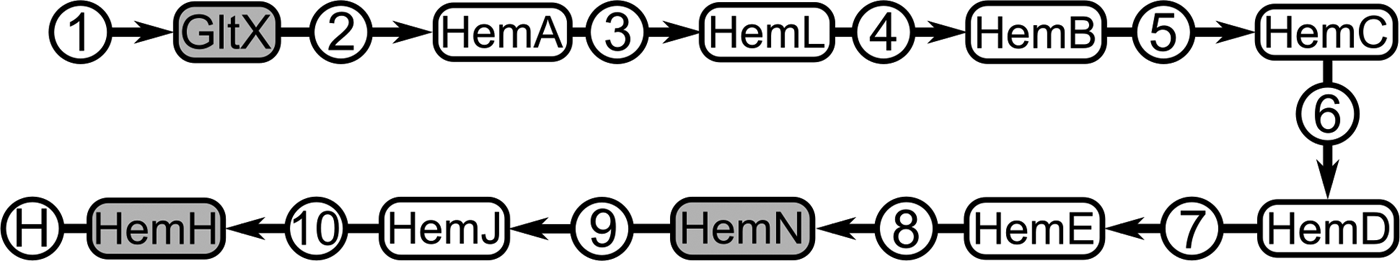
Fig. 3. Schematic representation of the haem-synthesis pathway in Ca. Kinetoplastibacterium spp. Compounds are represented within circles, and enzymes are within rounded-corner rectangles. Grey background highlights the enzymes for which genes were found in the Ca. K. sorsogonicusi genome. Compounds are 1: L-glutamate; 2: L-glutamyl-tRNA; 3: glutamate-1-semialdehyde; 4: aminolevulinic acid; 5: porphobilinogen; 6: hydroxymethylbilane; 7: uroporphyrinogen III; 8: coproporphyrinogen III; 9: protoporphyrinogen IX; 10: protoporphyrin IX; H: haem. Enzymes are GltX: glutamyl-tRNA synthetase (EC:6.1.1.17); HemA: glutamyl-tRNA reductase (EC:1.2.1.70); HemL: glutamate-1-semialdehyde 2,1-aminomutase (EC:5.4.3.8); HemB: aminolevulinic acid dehydratase (EC:4.2.1.24); HemC: porphobilinogen deaminase (EC:2.5.1.61); HemD: uroporphyrinogen III synthase (EC:4.2.1.75); HemE: uroporphyrinogen III decarboxilase (EC:4.1.1.37); HemN: oxygen-independent coproporphyrinogen-III oxidase (EC:1.3.99.22); HemJ: protoporphyrinogen oxidase (EC:1.3.3.4); HemH: ferrochelatase (EC:4.99.1.1).
We have also searched the unpublished draft sequence of the trypanosomatid host, K. sorsogonicus, for the haem-synthesis-specific genes, but found none.
Other biosynthetic pathways that have been previously shown, in other Ca. Kinetoplastibacterium spp., to be provided by the bacterium to complement the trypanosomatid host's metabolism were examined in Ca. K. sorsogonicusi and compared to other bacteria from the genus (Supplementary File 2). Pathways for the synthesis of essential amino acids as well as vitamins B6, pantothenic acid, folic acid and riboflavin are essentially identical between Ca. K. sorsogonicusi and the other Ca. Kinetoplastibacterium spp. compared.
Haem requirement test
In the first passage, the growth of K. sorsogonicus in culture was observed in both M199+ and M199− media. However, in the latter case, it was much slower. Taking into account potential leftovers of haemin on the cell surface or in intracellular depots, we passaged the trypanosomatids from M199− to new tubes containing both variants of media. It this case, the growth was observed only in M199+, whereas in M199− it stopped and after one week all cells in this medium were dead.
Discussion
Given a number of characteristic morphological features as well as the presence of the beta-proteobacterial endosymbiont (Votýpka et al. Reference Votýpka2014), Kentomonas is definitely a member of the subfamily Strigomonadinae. As mentioned above, in this subfamily the hosts and their respective endosymbionts almost always present (with the exception being A. ambiguus) a perfect pattern of co-speciation (Teixeira et al. Reference Teixeira2011). However, in the first description of the Kentomonas genus, the trypanosomatid host and its endosymbiont had discordant phylogenetic positions, as judged by the inference using SSU rRNA genes. Kentomonas was the earliest branch within its subfamily, while Ca. K. sorsogonicusi, albeit with low support values, appeared as the sister taxon to the endosymbionts of Angomonas spp. Thus, the bacteria from Strigomonas spp. seemed to be the first diverging lineage within the genus Ca. Kinetoplastibacterium. Our multigene phylogenetic analyses of 506 protein sequences of the endosymbiont, including Ca. K. sorsogonicusi, the five previously sequenced Ca. Kinetoplastibacterium genomes, and two other Alcaligenaceae (Achromobacter arsenitoxydans and Taylorella equigenitalis) as outgroups has shown with strong support that the Kentomonas endosymbiont is indeed at the base of the tree for this genus. Thus, the phylogenies of the host and endosymbiont agree, leaving A. ambiguus as the only exception to the strict co-speciation between Strigomonadinae and Ca. Kinetoplastibacterium spp.
As recently shown (Alves et al. Reference Alves2013a; Motta et al. Reference Motta2013), the members of the genus Ca. Kinetoplastibacterium have highly reduced genomes in comparison with other bacteria from the same family. For instance, Taylorella, a parasite of the genital tract of horses, demonstrates a genome more than twice as big as those of Ca. Kinetoplastibacterium spp., whereas in Achromobacter, a typically free-living bacterium, the genome is around nine times larger. In the current work, we have characterized the most reduced Ca. Kinetoplastibacterium genome found to date, that of Ca. K. sorsogonicusi. With the length of 741 697 bp, it is about 10% (70–90 kbp) shorter than other known genomes within this genus (810 172–833 125 bp). No obvious explanation for the extra loss of sequence presents itself, given that Kentomonas and its endosymbiont conceivably possess a similar life cycle and overall environment as Angomonas and Strigomonas. Further in vivo and in vitro studies of this subfamily, and in particular this new genus, might shed light on the possible reasons for the differences seen here.
In spite of genome size reduction, with loss of many genes that are essential for free-living and sometimes even parasitic bacteria, the genomes of endosymbionts preferentially retain genes encoding proteins involved in the interaction with the trypanosomatid host, as typically seen in other endosymbiotic relationships (Nowack and Melkonian, Reference Nowack and Melkonian2010). Overall, analysis of the predicted metabolic maps shows that this also may be the case in Ca. K. sorsogonicusi, with a sole exception (see below).
As seen in the comparative analyses between other Kinetoplastibacterium genomes, gene loss in Ca. K. sorsogonicusi has been spread throughout the genome, without obvious hot-spots of missing sequence (Fig. 1). Most gene losses observed do not significantly affect whole metabolic pathways, with the pointed exception of the haem-synthesis. As characterized in many biochemical and cell biology experiments over several decades (Chang et al. Reference Chang, Chang and Sassa1975; de Souza and Motta, Reference de Souza and Motta1999), and more recently by genomic methods (Alves et al. Reference Alves2011; Reference Alves2013b; Klein et al. Reference Klein2013; de Azevedo-Martins et al. Reference de Azevedo-Martins2015), the endosymbiont provides the trypanosomatid host with a number of important compounds such as haem, essential amino acids, vitamins, and lipids. Accordingly, as seen in the comparative metabolic analyses, the Kentomonas endosymbiont possesses many such genes. The only exception is the synthesis of haem from glutamate (the C5 or Beale pathway), for which almost all genes were lost. The first enzyme in this pathway, glutamyl-tRNA synthetase (gltX), is not specific for the pathway and participates in protein synthesis by ligating L-glutamate to the corresponding tRNAs, its presence being therefore unsurprising. The other two retained genes (hemN and hemH) are specific to the haem-synthesis pathway.
Haem plays a very important role in the metabolism of virtually all cellular organisms, especially the aerobic ones, being a co-factor used by a number of haem proteins (Panek and O'Brian, Reference Panek and O'Brian2002; Mense and Zhang, Reference Mense and Zhang2006). Accordingly, regular trypanosomatids obtain haem or one of its precursors from their hosts, whereas most Strigomonadinae, as well as the unrelated bacteria-containing trypanosomatid Novymonas esmeraldas, consume the haem produced by their endosymbionts (Kořený et al. Reference Kořený, Oborník and Lukeš2013; Kostygov et al. Reference Kostygov2016; Reference Kostygov2017; Horáková et al. Reference Horáková2017).
The genetic makeup of these organisms reflects their need for haem. Some trypanosomatids (from subfamilies Leishmaniinae and Strigomonadinae), possess the final three proteins of the haem-synthesis pathway; others (such as Phytomonas and Herpetomonas) contain only the last one, namely ferrochelatase; and, finally, Trypanosoma lost all the haem-synthesis-related genes (Kořený et al. Reference Kořený, Oborník and Lukeš2013).
Of the haem-synthesis-specific genes, the Kentomonas endosymbiont has apparently kept only those coding for the oxygen-independent enzymes coproporphyrinogen oxidase III (hemN) and ferrochelatase (hemH). The function of hemN in this organism is unclear, given that it is an enzyme restricted to the haem-synthesis pathway, to the best of our knowledge. The hemH enzyme, on the other hand, can be used to add iron to (or remove it from) the protoporphyrin ring. Our search of the draft nuclear genome of K. sorsogonicus has not revealed any of the haem-synthesis-specific genes or even their fragments. Thus, the genomic situation for this organism is most similar to Trypanosoma spp. In vitro, our haem requirement experiment confirmed the genomic inference, demonstrating that K. sorsogonicus is unable to grow in the absence of a haem source. Hence, haem is an essential compound for this organism.
Although there are very few known organisms that do not require haem, the trypanosomatid Phytomonas is of particular interest. This plant parasite has been recently demonstrated as the first and so far the only eukaryote that is able to live in the absence of haem (Kořený et al. Reference Kořený2012). The only cellular process in which haem is necessary in Phytomonas is the synthesis of ergosterol, a component of the plasma membrane that is not obligatory for the cell's viability (its precursor, lanosterol, is used in its stead). Interestingly, Phytomonas still possesses the hemH gene that is lacking from K. sorsogonicus. However, the latter organism has been shown here to fail growing without a source of haem, and thus must acquire the compound from its insect host, as the other trypanosomatids routinely do. Therefore, Phytomonas continues to be the only known eukaryote that can propagate in culture without any source of haem. The loss of haem-synthesis in the Kentomonas system suggests that either the extra haem provided to Strigomonas and Angomonas by their endosymbionts might be dispensable (or essential) only for these two genera or that there is some particularity of Kentomonas that makes haem less critical to its survival and reproduction, allowing it to thrive just with the haem provided by its invertebrate host, as is the case with almost all other trypanosomatids.
In the absence of more detailed information on the ecological and physiological conditions faced by K. sorsogonicus and its endosymbiont, it is difficult to make any functional and evolutionary interpretation of the loss of the haem biosynthesis pathway in these organisms. We can only speculate about this based on comparative data. Kinetoplastids do not possess this pathway, likely because of the toxicity of haem precursors. The loss of haem synthesis is considered advantageous for many parasites (as reviewed in Kořený et al. Reference Kořený, Oborník and Lukeš2013). The case of Kentomonas highlights the plasticity and adaptability of parasite genomes, which may change in response to environmental cues. The common ancestor of Strigomonadinae gained haem synthesis through endosymbiosis. While the bacterial symbionts of Angomonas and Strigomonas kept this capability, those of Kentomonas, which separated early from the rest of the subfamily, lost it. Such evolutionary plasticity, which is further exemplified by the ability of Phytomonas to live completely without haem, demonstrates that it is possible to lose, regain, and lose again the metabolic pathways for compounds currently considered essential.
For long, it has been a standard procedure in cultivating trypanosomatids isolated from insects to remove haemin from the culture medium; continued growth was considered as an evidence of endosymbiont presence. However, as we demonstrate here, despite bearing the intracellular bacterium, K. sorsogonicus cannot grow in culture without haem. Thus, the traditional test cannot be taken as a reliable criterion of the absence or presence of endosymbionts in trypanosomatid flagellates.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S003118201800046X
Acknowledgements
We thank members of our labs for stimulating discussions.
Financial Support
This work was supported by grants #2013/14622-3 São Paulo Research Foundation (FAPESP) to JMPA; ERC CZ (LL1601) award to JL; the Czech Grant Agency grant 16-18699S to JL and VY; RFBR grant 18-04-00138_A to AK and by ERD Funds, project OPVVV 16_019/0000759 to AK, VY and JL.
Conflict of Interest
None.
Ethical Standards
Not applicable.






