INTRODUCTION
Almost half of the world's population is at risk of developing malaria. Each year, an estimated 300 million people contract the disease and at least 1 million, mainly children, die (Sachs and Malaney, 2002). Plasmodium falciparum is the most widespread and virulent Plasmodium species infecting man. Annual mortality rates have been steadily rising and one of the most important factors is the increased resistance of P. falciparum to existing chemotherapeutic drugs. This realization has intensified efforts to identify new drugs and drug targets. DNA helicases play key essential roles in such biological processes as DNA replication, repair, recombination and transcription, by working in association with other proteins as part of a complex machine (Bachur et al. 1992). Helicases separate both strands of duplex DNA by breaking the hydrogen bonds formed between the bases. The energy to drive this reaction is derived from hydrolysis of nucleoside triphosphates. Helicases are classified according to their unwinding directionality, either 5′ to 3′ or 3′ to 5′, with respect to the strand they bind to, and along which they move.
DNA helicases are ubiquitous, having been identified in various prokaryotes and eukaryotes as well as in bacteriophages and viruses, and most of these organisms encode multiple helicase forms (Matson and Kaiser-Rogers, 1990). The anthracycline antibiotics that stabilize double-stranded DNA primarily by their intercalative binding show inhibitory effects on helicases from E. coli, SV40, and HeLa cells (Bachur et al. 1992, 1993). According to their amino acid sequences, several putative malaria parasite DNA helicases have been identified, based mostly on the canonical DEAD-,DEXH- or DEAH-box motif that is involved in the binding of ATP. Recently, an eIF-4A homologue that is the prototype of DEAD-box helicase from Plasmodium cynomolgi (PcDDH45) and ‘DEAD-box’ helicase (PfDH60) from Plasmodium falciparum have been cloned, expressed and characterized (Tuteja et al. 2002; 2003; Pradhan et al. 2005a,b). PfDH60 is a homologue of p68, present in various species including humans, and unwinds dsDNA in both directions (Pradhan et al. 2005b). However, the identification of malaria parasite helicases cannot be based solely on the sequence homology these enzymes may have with the cognate enzyme from E. coli, as many proteins may bind ATP but do not necessarily unwind DNA. Purification of helicases from organisms by monitoring the unwinding activity of the enzymes provides a better strategy for identifying both canonical and non-canonical helicases.
In this study, a novel 3′-5′ DNA helicase was isolated and purified from crude extracts of P. falciparum by using fast protein liquid chromatography (FPLC) and further characterized for its preferred substrates and requirements for DNA unwinding activity. The effects of anthracycline antibiotics on inhibition of P. falciparum helicase unwinding activity and parasite growth were also investigated.
MATERIALS AND METHODS
Parasites
Plasmodium falciparum K1, a chloroquine- and pyrimethamine-resistant strain from Thailand (Thaithong et al. 1983), was cultivated using a large-scale culture method (Chavalitshewinkoon and Wilairat, 1991). Parasite culture was started with 1% initial parasitemia after synchronization by sorbitol treatment (Lambros and Vanderberg, 1979). Parasites consisting principally of the schizont stage were harvested when parasitaemia reached 20–30%. The parasites were released from red cells by saponin lysis and parasite pellets were kept at −80 °C before use.
Preparation of P. falciparum crude extract
Parasite pellets were pooled (4·5 ml) and resuspended in 5 volumes of extraction buffer (50 mM Tris HCl, pH 7·6, 1 mM EDTA, 2 mM dithiothreitol (DTT), 0·01% Nonidet P40 (NP40), 1 mM phenylmethylsulfonyl fluoride (PMSF)). Parasites were broken by Dounce homogenization and an equal volume of dilution buffer (50 mM Tris HCl, pH 7·6, 1 mM EDTA, 2 mM DTT, 20% (w/v) sucrose, 0·01% NP40, 1 mM PMSF) was added. The parasite nuclei were lysed by the slow addition of 3 M KCl, until a final concentration of 0·5 M KCl was obtained. After stirring for 30 min on ice, the suspension was centrifuged at 100000 g for 40 min at 4 °C. The supernatant was precipitated by slowly adding ammonium sulfate (final concentration 0·35 g/ml) (55% ammonium sulfate) with constant stirring, and the mixture was stirred for 2 additional hours. The precipitate was collected by centrifugation at 3000 g for 40 min. The pellet was resuspended and dialysed against buffer A (25 mM Tris HCl, pH 8·0, 1 mM EDTA, 1 mM PMSF, 1 mM DTT, 5% sucrose, 20% glycerol, 0·01% NP40)(Fraction I). The supernatant was further precipitated by slowly adding ammonium sulfate to a final concentration of 0·5 g/ml (80% ammonium sulfate) and the pellet was resuspended and dialysed against buffer A (Fraction II).
Fraction I was loaded onto a 6 ml Resource S column. The column was washed with a 5-column volume of the same buffer and proteins were eluted with a 60 ml linear gradient (0–1 M KCl in buffer A) at a flow rate of 2 ml min−1, and 2-ml fractions were collected. Two peaks of helicase activity were obtained. The first peak of active fractions was eluted at 0·08–0·19 M KCl and the second peak at 0·28–0·48 M KCl. Purification of DNA helicases from these 2 peaks is in progress. Here, we describe the purification of DNA helicase from Fraction II.
Purification of P. falciparum 3′-5′ DNA helicase
All further purification steps were performed at 4 °C using an FPLC apparatus (Pharmacia). The dialysed protein from Fraction II was loaded onto a Resource Q column (6 ml, Pharmacia) equilibrated with buffer A. The column was washed with 54 ml of buffer A and proteins were eluted with 60 ml of linear gradient (0–1 M KCl in buffer A) at a flow rate of 2 ml min−1. Two ml fractions were collected and measured for DNA helicase activity as described below.
The active fractions were pooled and dialysed against buffer B (20 mM sodium phosphate buffer, pH 7·6, 1 mM EDTA, 1 mM PMSF, 1 mM DTT, 5% sucrose, 20% glycerol, 0·01% NP40). The proteins were loaded onto a Hitrap heparin column (5 ml, Pharmacia). The column was washed with 25 ml of buffer B and eluted with 40 ml linear gradient (0–1 M NaCl in buffer B) at a flow rate of 1 ml min−1. The eluates were collected into 27 fractions, each of 1·5 ml, and assayed for DNA helicase activity. Fractions containing helicase activity were pooled and dialysed against buffer C (buffer adjusted to pH 7·5) and applied to a Mono S column (1 ml, Pharmacia). The column was washed with 30 ml of buffer C and eluted with 8 ml linear gradient (0–1 M KCl in buffer C) at a flow rate of 0·25 ml min−1. Fractions of 0·25 ml were collected and assayed for DNA helicase activity. The active fractions were pooled and diluted to 50 mM KCl in buffer C. The diluted solution was loaded onto a single-stranded (ss)DNA column (1 ml, Pharmacia). The column was washed with 10 ml of buffer C and the enzyme was eluted with 8 ml of linear gradient (0–1 M KCl in buffer C) at a flow rate of 0·25 ml min−1, and 0·2 ml fractions were collected.
Preparation of 5′-labelled helicase substrate
All oligodeoxynucleotides were synthesized by the BioService Unit, National Science and Technology Development Agency, Thailand. The DNA substrate used in the helicase assay consisted of 32P-labelled oligodeoxynucleotide 1 (17 mer, 5′-GTAAAACGACGGCCAGT-3′) annealed to single-stranded M13mp19 (+) DNA (Invitrogen) to create a partial duplex (Tuteja et al. 1990). A total of 0·1 μg of 17-mer was labelled at the 5′-end with T4 polynucleotide kinase and [γ-32P] ATP and purified by precipitation in absolute ethanol. The radio-isotope labelled oligodeoxynucleotide was then annealed with 2·5 μg of single-stranded circular M13 mp19 DNA in 20 mM Tris-HCl, pH 7·5, containing 10 mM MgCl2, 100 mM NaCl and 1 mM DTT, by first heating at 95 °C for 1 min, transferring immediately to 65 °C for 2 min, and then cooling slowly to room temperature for 30 min. Non-hybridized oligonucleotide was removed by 2 passages through a 1 ml MicroSpin S-400 column (Pharmacia) followed by passage through an AutoSeq G-50 column (Pharmacia).
DNA helicase assay
DNA helicase was assayed under conditions as previously described (Tuteja et al. 1990, 1993, 1995) by measuring the unwinding of α-32P labelled partial duplex DNA molecule. The reaction mixture (10 μl) containing 2 μl of enzyme fraction, 20 mM Tris-HCl, pH 9·0, 8 mM DTT, 2 mM MgCl2, 2 mM ATP, 10 mM KCl, 4% (w/v) sucrose, 80 μg/ml BSA, and 1000 cpm of α-32P labelled helicase substrate was incubated at 37 °C for 20 min. The reaction was terminated with 10 μl of loading dye (10% Ficoll, 50 mM EDTA, 10 mM Tris-HCl, 0·25% xylene cyanol, 0·25% bromophenol blue). The unwound DNA product was separated from the duplex substrate by electrophoresis in a 20% non-denaturing polyacrylamide gel in 0·5× Tris-borate-EDTA buffer at 92 volts for 1·5 h. The gel was exposed to X-ray film to locate the unwound isotope-labelled fragment, which was then excised from the gel and submitted to Cerenkov counting.
One unit of DNA helicase activity is defined as the amount of enzyme that can unwind 1% of the DNA duplex substrate in 1 min at 37 °C.
Preparation of blunt-ended duplex DNA substrate
Oligodeoxynucleotide 2 (50 ng of 41-mer; 5′-AATTCGAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTG-3′) was 5′-end labelled with T4 polynucleotide kinase and [γ−32P] ATP. This labelled fragment was annealed with an equal amount of oligodeoxynucleotide 3 (41mer; 5′-CAGGTCGACTCTAGAGGATCCCCGGGTACCGAGCTC GAATT-3′) in 40 mM Tris-HCl, pH 7·5, 20 mM MgCl2 and 50 mM NaCl. The mixture was heated at 65 °C for 2 min and left to stand at room temperature for 30 min. Finally, the mixture was passed through a 1 ml MicroSpin S-400 column (Pharmacia) to remove free oligonucleotides.
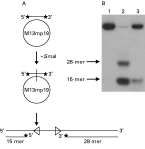
Determination of directionality of P. falciparum DNA helicase
Oligodeoxynucleotide 4 (50 ng of 42-mer; 5′-TCGAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTGCAGG-3′) was 5′-labelled using T4 polynucleotide kinase and [γ-32P] ATP and annealed to M13mp19 (+) DNA by using the modified methods of Tuteja et al. 1990. The partial duplex was passed twice through a 1 ml MicroSpin S-400 column (Pharmacia) and once through an AutoSeq G-50 column (Pharmacia) to remove unreacted radionucleotides. The annealed DNA was then labelled at the 3′-end with 10 μCi [ α−32P] dCTP and 5 units of DNA polymerase I (Klenow Fragment, BioLabs) at 23 °C for 20 min. After that, the concentration of dCTP was increased to 50 mM and the solution further incubated at 23 °C for 20 min. The unreacted nucleotides were removed by passing through a 1 ml MicroSpin S-400 column (Pharmacia). The annealed duplex was digested with 20 units of SmaI (BioLabs) at 25 °C for 1 h.
Unwinding in the 5′-to-3′ direction was detected by monitoring in 20% non-denaturing polyacrylamide gel the release of radioactive 28-mer, whereas unwinding in the 3′-to-5′ direction was observed by the release of the radioactive 15-mer from the substrate (Tuteja et al. 1990).
Preparation of fork-like DNA substrate
Oligodeoxynucleotide 5 [50 ng of 60-mer; 5′-(T)10CGAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTGCA(T)11-3′] was 5′-labelled, annealed and digested as described above. The partial duplex of M13-31 mer with hanging tail at 3′ or 5′ end substrate was prepared by using oligonucleotide 6 (50 ng of 44-mer; 5′-TCGAGCTCGGTACCCGGGGATCCTCTAGAG(T)14-3′ and oligodeoxynucleotide 7 (50 ng of 45-mer; 5′-GTTCCAGCGCTAGCTTCGAGCTCGGTACCCGGGGATCCTCTAGAG-3′), respectively. Oligodeoxynucleotides 6 and 7 were labelled at the 5′-end with T4 polynucleotide kinase and [γ-32P] ATP and precipitated by absolute ethanol, whereas free nucleotides were removed by washing twice in 70% ethanol. The labelled oligodeoxynucleotide was then annealed with 1·25 μg of single-stranded circular M13mp19 (+) strand DNA in 20 mM Tris-HCl (pH 7·5), 10 mM MgCl2, 100 mM NaCl and 1 mM DTT by heating at 95 °C for 1 min, transferring immediately to 65 °C for 2 min, and then cooling slowly to room temperature for 30 min. Non-hybridized oligodeoxynucleotides were removed by gel filtration through 1 ml of MicroSpin S-400.
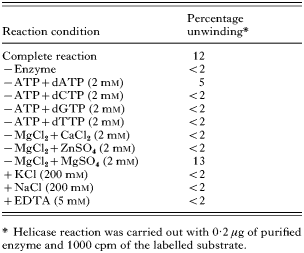
Reaction requirements of P. falciparum 3′-5′ DNA helicase unwinding activity
Helicase unwinding assays were performed as described above in the complete reaction, except for the changes of some ions or dNTPs shown in Table 2. The energy requirement was provided using 2 mM of dNTPs instead of 2 mM ATP in the complete reaction. The divalent cation requirement was tested by using 2 mM CaCl2, ZnSO4 and MgSO4 compared with 2 mM MgCl2 in the complete reaction. The presence of 5 mM EDTA in the complete reaction was also tested. The effect of the salt was evaluated using 200 mM KCl and NaCl.
Determination of molecular weight by SDS-PAGE
DNA helicase (2·0 μg) eluted from the ssDNA column (fraction 12–16) was mixed with 1× loading dye (62·5 mM Tris HCl, pH 6·8, 10% (v/v) glycerol, 2% (w/v) SDS, 5% (v/v) mercaptoethanol, 0·001% (w/v) bromophenol blue) and heated at 95 °C for 5 min. The solution was electrophoresed in 12% SDS-polyacrylamide gel and the protein band visualized by silver staining and compared with protein molecular weight markers (Fermentas) in the range 14·4–116·0 kDa.
Effects of anthracycline antibiotics on P. falciparum 3′-5′ DNA helicase activity
Aclarubicin, daunorubicin, doxorubicin, and nogalamycin were purchased from Sigma. These drugs were dissolved in dimethylsulfoxide (DMSO) to make stock solutions of 10−2 M that were stored at −20 °C. Drugs were serially diluted in 10 mM Tris-HCl, pH 9. The drugs were pre-incubated with 17-mer partial duplex DNA substrate (1000 cpm) for 10 min at 4 °C prior to addition of 0·2 μg purified 3′-5′ P. falciparum DNA helicase. The highest concentration of DMSO (0·2%) in the assay solution had no effect on DNA helicase activity.
In vitro sensitivity test of P. falciparum to anthracycline antibiotics
In vitro drug assays were performed in 96-well tissue culture plates (Nunc). A 1% parasitaemia of ring-stage P. falciparum K1 was used. A vol. of 25 μl of the drugs was added to 200 μl of erythrocyte suspension at 10-fold dilutions, starting from a maximum concentration of 9×10−4 M, to a minimum of 9×10−8 M. The highest concentration of DMSO (0·1%) had no effect on parasite growth. Cultures were incubated at 37 °C for 24 h under 5% CO2, and then 0·25 μCi of [3H] hypoxanthine (6·2 Ci/mmol, Amersham) were added to each well and the cultures were incubated for another 24 h. Erythrocytes were harvested using an automated sample harvester (Nunc), and radioactivity incorporated into the parasites counted in a liquid scintillation counter (Beckman). The experiments were triplicated. IC50 is defined as the concentration of drug inhibiting incorporation of [3H] hypoxanthine into parasites by 50%.
RESULTS
Purification of P. falciparum 3′-5′ DNA helicase
Large-scale cultivation of P. falciparum was essential for high parasite yield. Parasites were harvested mostly at schizont stages from cultures with 20% parasitaemia. Approximately 1 ml of packed parasites was obtained from 1 litre of culture. Enzyme purification, starting from 4·5 ml of parasite pellet, was approximately 1×1011 parasites. We have initiated purification of P. falciparum DNA helicase from its crude extract, and at least 3 molecular species based on their different fractionation properties were observed. One of them has been purified and characterized, whereas purification of the other helicases is in progress. The results of purifying P. falciparum 3′-5′ DNA helicase (3′-5′ PfDH) using FPLC are summarized in Table 1. Fraction II (7 ml), from 80% ammonium sulfate pellet, was loaded onto a Resource Q column. The active fractions (fraction numbers 12–17, Fraction III) were eluted at 0·34–0·52 M KCl in buffer A. Fraction III was dialysed against buffer B and applied onto a Hitrap heparin column (5 ml) equilibrated with buffer B. The active fractions (fraction numbers 12–15, Fraction IV) were eluted at 0·37–0·48 M NaCl in buffer B. Fraction IV, following dialysis against buffer C, was loaded onto a Mono S column and active fractions (fraction numbers 13–15, Fraction V) were eluted at 0·28–0·37 M KCl in buffer C. Fraction V, diluted to 50 mM KCl in buffer C and applied onto an ssDNA column, yielded active fraction VI (fraction numbers 12–16, eluting at 0·1–0·22 M KCl in buffer C) (Fig. 1).

Fig. 1. Unwinding activity of Plasmodium falciparum DNA helicase (PfDH A) eluted from the ssDNA column. The standard displacement assay with 17 mer/M13 partial duplex substrate and 2 μl of enzyme from each fraction was performed as described in the Materials and Methods section. The number above the figure is the fraction eluted from the column.

From 4·5 ml of parasite pellet, only 96 μg of purified DNA helicase was recovered, with a specific activity of 3210 units/mg protein. The enzyme was relatively unstable and lost 25% of its activity following storage at −70 °C for 6 months. Purified P. falciparum DNA helicase (Fraction VI) was free of nuclease, DNA polymerase and DNA topoisomerase (data not shown). Fraction V showed 1 band of 90 kDa on SDS-polyacrylamide gel electrophoresis (Fig. 2).

Fig. 2. Determination of molecular weight of PfDH A by SDS-PAGE. The protein was separated in 12% SDS-polyacrylamide gels and visualized with silver staining. Lane 1 shows size markers indicated in kDa. Lane 2 is 2·0 μg of pooled active fractions from the ssDNA column.
The reaction properties of 3′-5′ PfDH are shown in Table 2. The helicase reaction required the presence of ATP or dATP as cofactors, but dATP supported unwinding at only 42% of the efficiency of ATP. Statistical analysis using Poisson regression showed that dATP was statistically different from the negative control and the other tested conditions in Table 2 (P=0·011). Other dNTPs failed to support any detectable unwinding of the purified enzyme (Table 2). 3′-5′ PfDH A required divalent cations (Mg2+ but not Ca2+ or Zn2+) for its activity with a P value of 0·748 when MgSO4 is compared with the complete reaction with MgCl2. Enzyme activity was inhibited by adding 5 mM EDTA to the standard condition. The unwinding reaction using 0·2 μg of purified enzyme was linear for approximately 20 min and reached 20% completion in 90 min (Fig. 3). KCl and NaCl at a concentration of 200 mM abolished helicase activity.

Fig. 3. Time dependence of PfDH A helicase activity. Percentage unwinding was plotted against incubation time. The reaction contained 0·2 μg of PfDH A. H is heat-denatured 17 mer/M13 substrate.
Table 2. Reaction requirement of Plasmodium falciparum 3′-5′ DNA helicase unwinding activity (−, without; +, with.)

Since the circular partial DNA duplex used as substrate in the helicase assay does not allow determination of directionality, both 5′- and 3′-labelled linear substrates obtained by digestion of circular partial duplex substrate with SmaI were employed as substrates to determine enzyme directionality. Unwinding in a 5′-to-3′ direction should result in the release of radio-isotope-labelled 28-mer while unwinding in a 3′-to-5′ direction should release labelled 15-mer from the substrate. P. falciparum DNA helicase unwound only the 15-mer product and not the 28-mer from the linear substrate (Fig. 4), indicating that it moves only in a 3′-to-5′ direction along the DNA strand to which it binds. In addition, its 3′-to-5′ directionality was confirmed when the other two fork-like substrates were used to determine substrate preference (Fig. 5E and F). P. falciparum DNA helicase moved along the bound strand in the direction 3′ to 5′ and released a 24-mer product (Fig. 5E), but it did not unwind the 3′-labelled substrate constructed for testing for 5′-to-3′ directionality (Fig. 5F).

Fig. 4. Directionality assay and an autoradiogramme of radio-isotope labelled oligomer unwound from partial DNA duplex by purified PfDH A. A directionality assay schematic is shown (A). The linear partial duplex substrate used to determine the direction of PfDH A was prepared as described in the Materials and Methods section. Lane 1 is a control (reaction without enzyme), lane 2 is a heat-denatured substrate, and lane 3 is a reaction with 0·2 μg of purified PfDH A (B).

Fig. 5. Substrate specificity of Plasmodium falciparum 3′-5′ DNA helicase activity. Each panel shows the structure of substrate used, autoradiogramme of the gel, and percentage unwinding of the enzyme. Lane 1 is native substrate, lane 2 is substrate with enzyme, and lane 3 is heat-denatured substrate. Asterisk denotes the end of DNA labelled with 32P.
3′-5′ PfDH could unwind the 17-mer partial duplex substrate (Fig. 5A) but not 31-mer partial duplex substrate with hanging tail at 5′- or 3′-end (Fig. 5C and D). Linear substrates with 5′ and 3′ hanging tails made by SmaI digestion were also used to test for substrate preferences of the parasite enzyme (Fig. 5E and F). The results showed that 3′-5′ PfDH could use linear substrates and open up the double helix. Moreover, this confirmed that the purified enzyme moves in a 3′-to-5′ direction because 23% unwinding activity was found using 5′-labelled fork-like substrate (Fig. 5E), whereas <2% unwinding activity was observed with 3′-labelled fork-like substrate (Fig. 5F). The purified enzyme was unable to unwind blunt-ended duplex DNA substrate, as found in other eukaryotic DNA helicases (Fig. 5B). All 4 anthracycline antibiotics tested inhibited P. falciparum DNA helicase activity in the μM range, and daunorubicin was the most effective drug against the parasite enzyme, with an IC50 of 2 μM, whereas nogalamycin was the most potent drug for inhibiting parasite growth, with an IC50 of 0·1 μM (Table 3).

DISCUSSION
In this study, we report the existence of a novel P. falciparum 3′-to-5′ DNA helicase, which we have named P. falciparum DNA helicase A (PfDH A). PfDH A has an apparent molecular weight of 90 kDa, which is different from PfDH60, which was previously reported by Pradhan et al. (2005a). PfDH60 is a putative ATP-dependent RNA helicase encoded by the PfDH60 gene, which was obtained by using a ‘DEAD-box’ motif as query. Its deduced amino-acid sequence consists of 516 amino acid residues with a predicted molecular mass of 59·8 kDa, and it is a basic protein with a calculated isoelectric point of ~8·5. The PfDH60 gene is located on chromosome 12 of P. falciparum and its ‘PlasmoDB’ entry number is PFL1310c (Pradhan et al. 2005b). Based on calculation of the molecular weight, PfDH A consists of approximately 782·6 amino acids. Based on the database of P. falciparum, approximately 72 putative genes may encode for both DNA helicases and RNA helicases. Among them, at least 29 putative DNA helicase genes were found by using ‘DNA helicase’ as query but only 26 genes showing scores from Pfam database as shown in Table 4, and the product of PFI0910w, consisting of 728 amino acids, is the most likely gene to encode PfDH A because it has a molecular weight of about 85·5 kDa compared with the 90 kDa purified protein. Unfortunately, the result could not be confirmed either by amino-acid sequencing, which may be due to N-terminal blocking, or by mass spectrometry.

PfDH A was present in very low abundance: from approximately 95·83 mg of P. falciparum protein we recovered only 96 μg of purified enzyme. PfDH A was present in the 80% (w/v) ammonium sulfate precipitate, unlike human 3′-5′ DNA helicases, which are mainly present in the 55% (w/v) ammonium sulfate precipitate (Tuteja et al. 1990, 1992, 1993, 1995). The major difficulty in measuring helicase activity in the crude extract of eukaryotic cells is the abundance of nucleases that destroy labelled substrates (Thommes and Hubscher, 1992). Despite adding 1 mM PMSF, 20% glycerol and 5% sucrose at each enzyme purification step, PfDH A was still relatively labile, losing 25% of its activity after 6 months, even when stored at −70 °C.
DNA helicase unwinds duplex DNA and translocates along DNA in a process that is coupled to the binding and hydrolysis of nucleoside triphosphates. The DNA helicase activity of PfDH A was totally dependent on ATP hydrolysis. Other dNTPs failed to support the DNA unwinding activity of PfDH A. Replacement of ATP with dATP decreased PfDH A activity by approximately 50%, and its requirement was statistically different from other dNTPs (P=0·011), whereas 95% activity was demonstrated in PfDH60 (Pradhan et al. 2005a). The ATP and dATP requirements of PfDH A are similar to those of most human DNA helicases, except HDH VIII, which can use only ATP (Costa et al. 1999). Mouse helicase can use all four NTPs (Seki et al. 1987), and SV40 T antigen helicase uses dTTP (Goetz et al. 1988).
Helicase activity requires the presence of divalent cations as a cofactor in NTP hydrolysis. Mg2+ was essential for the unwinding activity of PfDH A, and could not be replaced with Ca2+ and Zn2+, a situation similar to most eukaryotic helicases (Zhang and Grosse, 1991). MgCl2 could be replaced by MgSO4 for the unwinding activity of PfDH A, which has also been found in HDH I and III (Tuteja et al. 1990, 1992). Since changes in salt concentration influence the stability and specificity of protein-DNA complexes (Lohman and Bjornson, 1996), the salt requirement of PfDH A was investigated. PfDH A was inactive in the presence of 200 mM KCl or NaCl, as was found in HDH II, V, and VI (Tuteja et al. 1993, 1994, 1995), PfDH 60 (Pradhan et al. 2005a), calf thymus (Matson and Bean, 1995), and SV40 T antigen helicases (Goetz et al. 1988), whereas HDH I was 90% active at this salt concentration (Tuteja et al. 1990).
DNA helicases exhibit specific polarity, which is defined as the direction of its movement on an initially bound ssDNA template. PfDH A unwinds unidirectionally in the 3′-to-5′ direction with respect to the DNA strand on which it is bound. An absence of 36-mer product in Fig. 5F indicates that PfDH A lacks 5′-to-3′ directionality. It is absolutely different from PfDH 60, which unwinds dsDNA in both directions (Pradhan et al. 2005b). PfDH A prefers a fork-like substrate, as it showed 23% unwinding activity on a fork-like substrate with an annealed portion of 16-mer compared with 12% of products from a short duplex with an annealed portion of 17-mer which is nearly the same annealed size. The requirement of a fork-like substrate has also been observed in both PcDDH 45 and PfDH 60 (Tuteja et al. 2003; Pradhan et al. 2005a). However, PfDH A could not unwind a fork-like structure on the substrate with an annealed 31-mer portion, indicating that PfDH A can unwind only short duplexes. PfDH 60 is also unable to unwind the longer duplex region of 32-mer (Pradhan et al. 2005b). Based on these results, neither P. falciparum DNA helicases are processive enzymes in vitro. Among the human 3′-5′ DNA helicases, HDH V showed complete unwinding activity only with short (17-mer) duplex DNA molecules, and is known to be a non-processive enzyme (Vindigni et al. 2001). In addition, PfDH A could not unwind a blunt-ended substrate, like most human DNA helicases, and this failure may be due to the requirement of helicase of an initial binding site composed of ssDNA (Matson and Bean, 1995). Only a few helicases are able to unwind fully duplex substrates; these include E. coli DNA helicase II (Runyon and Lohman, 1989) and large T antigen helicase of SV40 (Stahl and Knippers, 1987).
The purified PfDH A had some characteristics similar to HDH VI, such as 3′-to-5′ directionality, a fork-like substrate preference, unwinding of short duplex, a requirement for ATP, activity supported by Mg2+ but not Ca2+ and Zn2+, and salt inhibition by 200 mM KCl or NaCl. However, PfDH A differs from HDH VI in its lower binding affinity to ssDNA column and its lower molecular weight (HDH VI is 128 kDa) (Vindigni et al. 2001).
As DNA helicases play important roles in chromosomal replication, they provide suitable and potential targets for chemotherapy. No specific inhibitors of helicases have been found so far, however, inhibitory effects on helicase activity of anthracycline antibiotics have been reported (Bachur et al. 1992, 1993). Anthracyclines can also inhibit DNA polymerases (Chandra et al. 1972; Goodman et al. 1974), RNA polymerases (Hartmann et al. 1964), topoisomerase II (Tewey et al. 1984), DNA ligase (Ciarrocchi et al. 1991) and DNA repair enzymes (Lee et al. 1974). Our study showed that anthracyclines could inhibit the unwinding activity of PfDH A. The most potent inhibitor of enzyme activity was daunorubicin, with an IC50 of 2 μM, which indicates that PfDH A is more sensitive to this drug than both Plasmodium cynomolgi helicase, PcDDH45 (Tuteja et al. 2003), and human DNA helicase, HDH II (Bachur et al. 1993). On the other hand, nogalamycin was the most effective compound against parasite growth, with an IC50 of 0·42 μM. The results of this study showed no relationship between inhibition of helicase activity and parasite growth, since anthracyclines also inhibit other enzymes involved in DNA replication, as mentioned above. From the present work, it appears that PfDH A is present at the schizont stage of P. falciparum and differs from both human DNA helicases and previously expressed enzymes from P. falciparum and non-human malaria parasites. However, its function and roles in the DNA replication of parasites require elucidation, to explore its potential as a new anti-malarial drug target.
This study was supported by the Thailand Research Fund (RSA/14/2543) and a grant from the Faculty of Graduate Studies, Mahidol University. The authors thank Professor Prapon Wilairat for helpful discussions and assistance in preparing the manuscript and Mr Paul R. Adams for critical reading of the manuscript.