



Introduction
The vicuña (Vicugna vicugna) is a wild South American camelid distributed in the high areas of the Andes, from Ecuador to the north of Argentina, covering areas of Peru, Bolivia and Chile (Wheeler, Reference Wheeler2012). The vicuña is one of the animals of the world that has its very fine fibre (Sahley et al., Reference Sahley, Vargas and Valdivia2007). Due to this, the vicuña was an animal threatened due to poaching. This illegal activity reduced the population of animals in the 1950s, reaching around 10 000 animals throughout South America. Strategies carried out to conserve the vicuña population in Peru have been successful (Wheeler, Reference Wheeler2012). Currently, the vicuña population is about 208 000 animals throughout Peru (Acebes, Reference Acebes and González2020). However, the increase in the vicuña population was accompanied by the appearance or increase of some diseases, such as sarcoptic mange (Gomez-Puerta et al., Reference Gomez-Puerta, Olazabal, Taylor, Cribillero, Lopez-Urbina and Gonzalez2013; Montecino-Latorre et al., Reference Montecino-Latorre, Napolitano, Briceño and Uhart2020).
Sarcoptic mange, caused by Sarcoptes scabiei, is a common infectious parasite disease in domestic and wild mammals (Pence and Ueckermann, Reference Pence and Ueckermann2002; Arlian and Morgan, Reference Arlian and Morgan2017). Infected animals manifest typical signs such as hair loss, dermal scabs and pruritus (Pence and Ueckermann, Reference Pence and Ueckermann2002). Currently, sarcoptic mange is documented in new geographic areas and new hosts, mainly wild mammals, and it is considered an emerging wildlife disease (Astorga et al., Reference Astorga, Carver, Almberg, Sousa, Wingfield, Niedringhaus, Van Wick, Rossi, Xie, Cross, Angelone, Gortazar and Escobar2018; Escobar et al., Reference Escobar, Carver, Cross, Rossi, Almberg, Yabsley, Niedringhaus, Van Wick, Dominguez-Villegas, Gakuya, Xie, Angelone, Gortazar and Astorga2021).
Sarcoptic mange is often considered an animal welfare issues, and can be a conservation issue in some circumstances [e.g. as seen in wolves, foxes, coyotes and wombats (Pence and Ueckermann, Reference Pence and Ueckermann2002; Fraser et al., Reference Fraser, Charleston, Martin, Polkinghorne and Carver2016; Astorga et al., Reference Astorga, Carver, Almberg, Sousa, Wingfield, Niedringhaus, Van Wick, Rossi, Xie, Cross, Angelone, Gortazar and Escobar2018)]. In South America, this disease is considered a severe problem for the vicuña population. Some studies showed up to 37% prevalence in vicuñas (Castillo et al., Reference Castillo, Cisneros, Gomez-Puerta and Gálvez-Durand2019), and the disease was also associated with the mortality of vicuñas in a Peruvian National Reserve (Korswagen, Reference Korswagen2016).
Molecular studies have provided much information about the epidemiology of this disease. Use of several gene loci such as internal transcribed spacer 2 (ITS2), ribosomal 12S and 16S RNA (rRNA) and mitochondrial cytochrome c oxidase subunit I (cox1) genes has helped to molecular typing of S. scabiei (Zahler et al., Reference Zahler, Essig, Gothe and Rinder1999; Walton et al., Reference Walton, Dougall, Pizzutto, Holt, Taplin, Arlian, Morgan, Currie and Kemp2004; Amer et al., Reference Amer, El Wahab, Metwaly Ael, Ye, Roellig, Feng and Xiao2014; Li et al., Reference Li, Sun, Xie, Zhou, Gu, Lai, Peng and Yang2018). These studies have shown specific genotypes of S. scabiei for some populations and species of mammals (Oleaga et al., Reference Oleaga, Alasaad, Rossi, Casais, Vicente, Maione, Soriguer and Gortazar2013; Amer et al., Reference Amer, El Wahab, Metwaly Ael, Ye, Roellig, Feng and Xiao2014). Likewise, the use of microsatellites has helped to give more precise and solid results regarding the mite population in the same host species or individual compared with ribosomal and mitochondrial markers (Oleaga et al., Reference Oleaga, Alasaad, Rossi, Casais, Vicente, Maione, Soriguer and Gortazar2013; Moroni et al., Reference Moroni, Angelone, Pérez, Molinar Min, Pasquetti, Tizzani, López-Olvera, Valldeperes, Granados, Lavín, Mentaberre, Camacho-Sillero, Martínez-Carrasco, Oleaga, Candela, Meneguz and Rossi2021).
The molecular epidemiology of sarcoptic mange in vicuñas remains unknown. This study represents a contribution to research on the sarcoptic mange prevalence in vicuñas from Southern Peruvian Andes and to the molecular characterization of Sarcoptes mites collected from vicuñas, analysing part of the cox1 gene and the ITS2 region.
Materials and methods
Ethical statement
This study was reviewed and approved by the Forest and Wildlife Service of Peru (SERFOR) (No 246-2018-DGGSPFFS).
Animals
During May and June of 2018, a total of 181 wild vicuñas were clinically evaluated for lesions compatible with mange during the shearing season in four localities (Chilca, Palccoyo, Phacco and Sibina Sallma) located in Canchis province in Cuzco, in the Southern Peruvian Andes, at an altitude between 4800 and 5200 m above sea level (Fig. 1). During this event, commonly known as the ‘chaku’, all animals were clinically evaluated by veterinarians to diagnose the presence of lesions compatible with mange. For this, areas of the face, ear, chest, abdomen, axillae, groin, legs and interdigital spaces were examined since the disease occurs mainly in these areas in vicuñas (Gomez-Puerta et al., Reference Gomez-Puerta, Olazabal, Taylor, Cribillero, Lopez-Urbina and Gonzalez2013). In addition, skin scrapings samples from each suspected animal were collected and stored in plastic vials with 70% ethanol duly labelled, indicating the animal data and affected area.
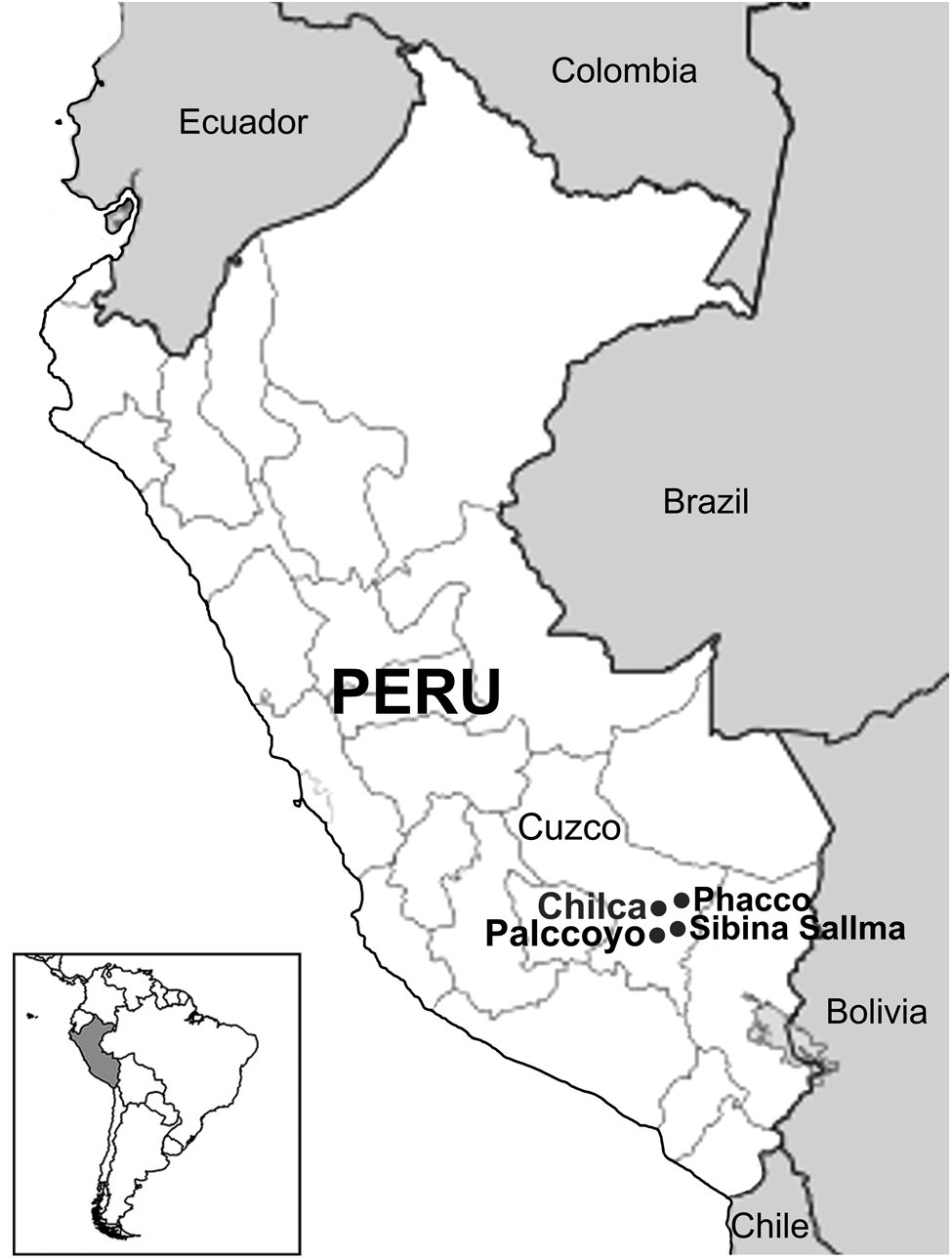
Fig. 1. Physical map of Peru and geographic location of the study site.
Microscopic diagnosis
Some tissues from skin scraping samples were digested in tampon buffer lysis (see below DNA extraction). The mites were collected under a stereomicroscope and then mounted in glass slides using Hoyer's medium (Krantz and Walter, Reference Krantz and Walter2009). The mites were examined morphologically under a microscope at 100× or 400× magnification, and identified using morphological keys previously described (Fain, Reference Fain1968).
DNA extraction
Some (n = 25) tissues (~3 mm3) from skin scraping samples were put in a 2 mL vial and dehydrated in a vacuum centrifuge. Then, 100 μL of tampon buffer lysis (40 mm Tris-HCl, 100 mm NaCl, 20 mm EDTA, 1% SDS, 0.2 mg mL−1 of proteinase K, pH 7.2) was added in each tube and was incubated at 56°C for 2–3 h. After this, mites were collected from each tube under a microscope (Carl Zeiss, Germany). DNA was extracted by an individual mite using Chelex method (Gomez-Puerta et al., Reference Gomez-Puerta, Alarcon, Pacheco, Franco, Lopez-Urbina and Gonzalez2016). DNA was extracted from 50 mites, two randomly selected mites from each scabietic vicuña.
PCR protocol and sequencing
A PCR was used to amplify approximately 421 base-pairs (bp) of the cox1 mitochondrial gene, using a protocol and primers (772 and 773) proposed by Navajas et al. (Reference Navajas, Gutierrez, Bonato, Bolland and Mapangou-Divassa1994). Also, the complete ITS2 region was amplified using primers LGF2 (5′-CGT TTT AAA TGC AAA ATT CAA-3′) and LGR2 (5′-GCC GTT ACT AAG GGA ATC-3′) designed in this study, and the following PCR protocol reaction: 4 μL of DNA, 10 μ m of each primer, 25 μL of Dream Taq Green PCR Master Mix (2×) (Thermo Fisher Scientific, USA) and ultrapure water in a final volume of 50 μL. Thermal cycler conditions were 94°C for 30 s, 45°C for 30 s and 72°C for 30 s, repeated 36 times. The PCR products were visualized by 1.5% agarose gel electrophoresis staining with ethidium bromide.
All PCR-positive samples, corresponding to 50 mites, were purified and sequenced using an ABI 3100 automated sequencer (Applied Biosystems, USA). The accuracy of the sequence was confirmed by bi-directional sequencing of two separate PCR products. The sequences were analysed using the software ChromasPro 2.1.8 (http://technelysium.com.au/wp/chromaspro). All sequences were compared with reference sequences from the GenBank using the Blast (https://blast.ncbi.nlm.nih.gov/Blast.cgi). The MEGA-X software (http://www.megasoftware.net/) was used to construct a phylogenetic tree according to the Maximum Likelihood method using the Tamura three-parameter model (Tamura, Reference Tamura1992; Kumar et al., Reference Kumar, Stecher, Li, Knyaz and Tamura2018). All sequences were subjected to 1000 bootstrap replications for the accuracy of the constructed phylogenetic tree. Unique nucleotide sequences of the cox1 gene and ITS2 region of S. scabiei from vicuñas were deposited in the GenBank under accession numbers MZ569438 to MZ569487 for cox1 and MZ574503 to MZ574550 for ITS2.
Data analysis
Sarcoptic mange diagnosis and demographic data (age, sex and geographical origin) were analysed as categorical variables. Age was categorized in younger and older than 1 year. The association of sarcoptic mange diagnosis and demographic variables was evaluated using the χ 2 test, considering 95% confidence intervals (95% CI). Data were analysed using Stata 15.0 (StataCorp, College Station, TX, USA).
Results
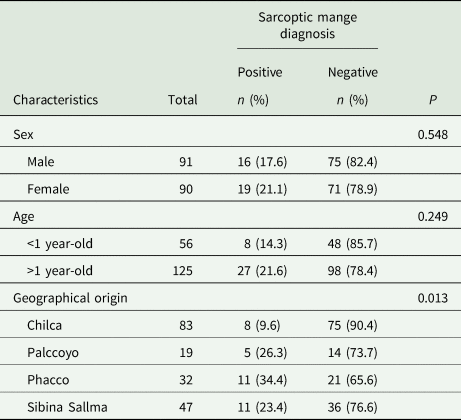
Lesions compatibles with mange were localized in chest, abdomen, axillae and groin zones (Fig. 2). Sarcoptic mange was suspected in 35 of 181 (19.3%; 95% CI 13.3–24.7%) vicuñas, leading to the collection of skin scrapings. Microscopic examination of the skin scraping samples confirmed the occurrence of S. scabiei in all of these samples. Infestation by S. scabiei did not differ between vicuñas sex and age (Table 1). Sarcoptic mange showed differences between localities, with low infestation rates from Chilca, relative to other locations (Table 1).

Fig. 2. Affected vicuñas showed skin lesions, mainly affecting internal legs (A), and chest and abdomen (B).
Table 1. Sarcoptic mange in vicuñas (Vicugna vicugna) from Cuzco in the Southern Peruvian Andes (n = 181) by sex, age and geographical origin as indicated by univariate analysis
DNA isolates were performed on 50 S. scabiei individual mites from 25 infected vicuñas (two mites per vicuña). A total of 50 partial nucleotide sequences (421 bp) of the cox1 gene were obtained from these samples, and the sequences had 94–99% similarity with previous S. scabiei sequences recorded in the GenBank. The S. scabiei cox1 sequences analysis from vicuñas showed two types of sequences with a single nucleotide polymorphism, a transition cytosine (C) by thymine (T). Regarding geographical origin, vicuñas from Chilca and Sibina Sallma were parasitized with the two types of S. scabiei cox1 sequences. Coinfection with both sequence types was not observed in any vicuña (see Supplementary Table S1). Phylogenetic analysis by the Maximum Likelihood method showed a cluster of S. scabiei from vicuñas. This cluster included S. scabiei cox1 sequences from canids (Fig. 3A).
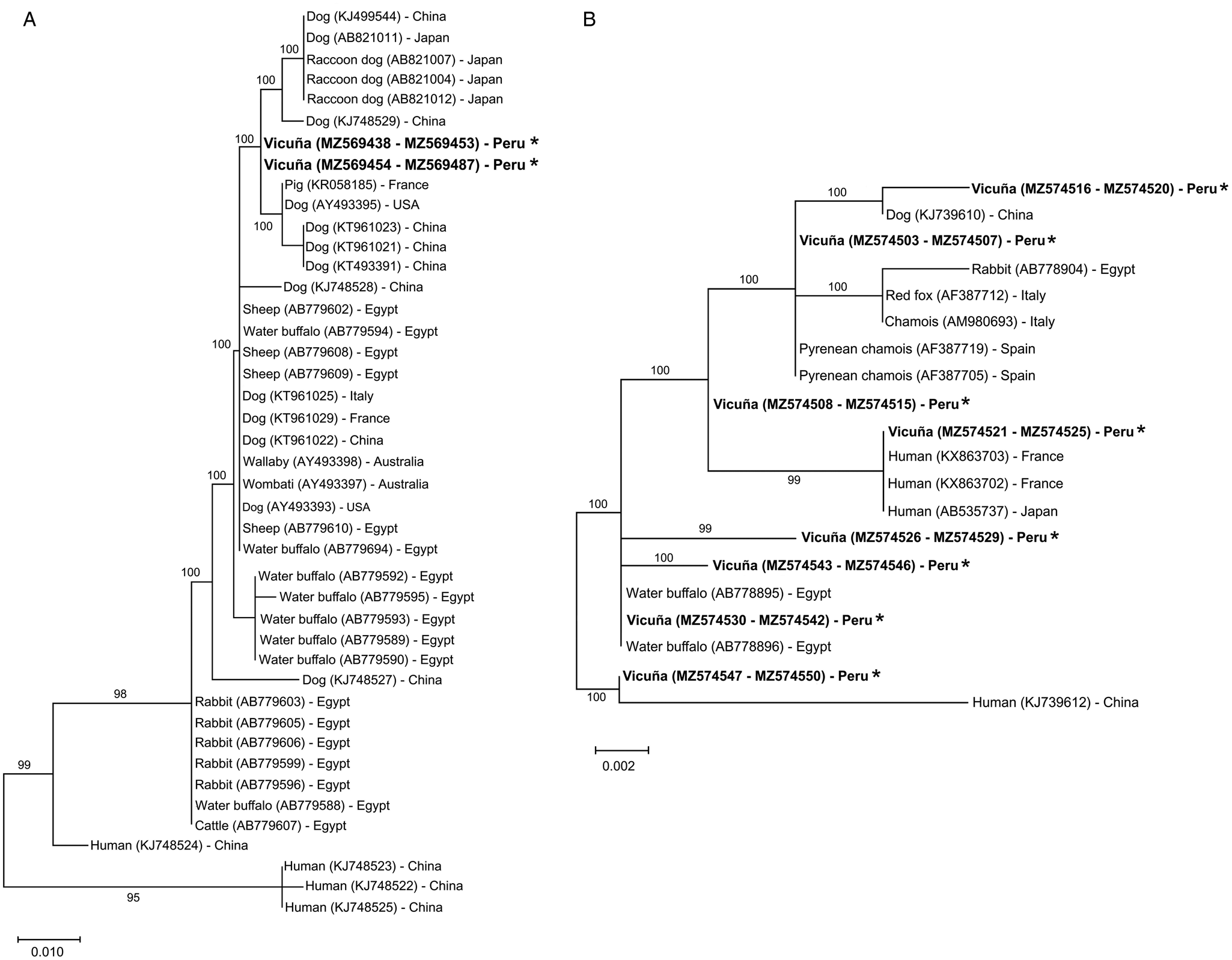
Fig. 3. Phylogenetic relationships of Sarcoptes scabiei collected from vicuñas (Vicugna vicugna) from Southern Peruvian Andes. Evolutionary analyses by the Maximum Likelihood method of the partial cox1 gene (A) and ITS-2 region (B) sequences were based on genetic distances calculated by the Tamura 3-parameter model. The percentage of trees in which the associated taxa clustered together is shown next to the branches. The trees were drawn to scale with branch lengths measured in the number of substitutions per site (scale bar for cox1 and ITS2 indicates 10 and 2 substitutions per 1000 nucleotide positions, respectively). This analysis involved 46 and 20 nucleotide sequences for cox1 and ITS2, respectively. All positions with <95% site coverage were eliminated, i.e. fewer than 5% alignment gaps, missing data and ambiguous bases were allowed at any position (partial deletion option). The sequence accession number is in parenthesis, followed by the origin country. Asterisk (*) indicates the sequences for this study.
The ITS2 region from 48 S. scabiei collected from 25 vicuñas was fully amplified and sequenced (total nucleotide sequence = 304 bp). The sequences were compared with previous S. scabiei sequences registered in the GenBank and showed an identity of 99.8–100%. A polymorphism was evidenced in the ITS2 nucleotide sequences of S. scabiei from vicuñas, showing a genetic variation of up to 1.4%. The polymorphic sites were represented by four transitions (C and T, alignment positions 14 and 18; A and G, alignment position 52 and 219), and five transversions (A and C, alignment positions 17 and 18; A and T, alignment positions 256 and 274; G and T, alignment position 294). This polymorphism in the ITS2 nucleotide sequences showed eight haplotypes for S. scabiei from vicuñas (Fig. 3B). Of these haplotypes, three were identical to those previously registered in the GenBank (Table 2, Supplementary Table S1). The remaining five corresponded to new S. scabiei haplotypes.
Table 2. Data information of the Sarcoptes scabiei isolated from vicuñas included in the molecular study

1 Localities from Canchis Province, Cuzco, in the Southern Peruvian Andes.
Discussion
This study indicates that sarcoptic mange is a common disease in vicuñas from Cuzco, in the Southern Peruvian Andes, showing a prevalence of 19.3%. Although epidemiological studies of sarcoptic mange in vicuñas are very limited, reports indicate prevalence ranging from 12 to 37% in Peru (Castillo et al., Reference Castillo, Cisneros, Gomez-Puerta and Gálvez-Durand2019; Gomez-Puerta et al., Reference Gomez-Puerta, Olazabal, Taylor, Cribillero, Lopez-Urbina and Gonzalez2013). Likewise, sarcoptic mange outbreaks have been documented in the neighbouring countries, reporting outbreaks in individual vicuñas from Chile (Montecino-Latorre et al., Reference Montecino-Latorre, Napolitano, Briceño and Uhart2020) and prevalence of 46.2% (6/13 animals) in Bolivia (Ruiz, Reference Ruiz2016) and 0.9% (4/450 animals) in Argentina (Arzamendia et al., Reference Arzamendia, Neder, Marcoppido, Ortiz, Arce and Vilá2012). As is known, sarcoptic mange is a parasite disease with characteristic and evident clinical signs in wildlife (Escobar et al., Reference Escobar, Carver, Cross, Rossi, Almberg, Yabsley, Niedringhaus, Van Wick, Dominguez-Villegas, Gakuya, Xie, Angelone, Gortazar and Astorga2021). In Peru, sarcoptic mange is an important disease in vicuña populations. Recently, a die-off occurred in vicuñas from the Tanta community located in the Paisajística Nor Yauyos Cochas Reserve in the Central Andes of Peru. Of the total number of dead vicuñas in this reserve, 64% were caused by sarcoptic mange (Korswagen, Reference Korswagen2016). This research is the first molecular study of S. scabiei from South American camelids in Peru. In addition, the study contributes new S. scabiei reference sequences from vicuñas.
Molecular tools are widely used to understand the epidemiology, host specificity and geographic separation of S. scabiei (Amer et al., Reference Amer, El Wahab, Metwaly Ael, Ye, Roellig, Feng and Xiao2014; Fraser et al., Reference Fraser, Shao, Fountain-Jones, Charleston, Martin, Whiteley, Holme, Carver and Polkinghorne2017; Li et al., Reference Li, Sun, Xie, Zhou, Gu, Lai, Peng and Yang2018). As shown in other studies, the S. scabiei cox1 genetic marker shows nucleotide sequence polymorphisms eventually related to their host (Walton et al., Reference Walton, Dougall, Pizzutto, Holt, Taplin, Arlian, Morgan, Currie and Kemp2004; Amer et al., Reference Amer, El Wahab, Metwaly Ael, Ye, Roellig, Feng and Xiao2014; Fraser et al., Reference Fraser, Shao, Fountain-Jones, Charleston, Martin, Whiteley, Holme, Carver and Polkinghorne2017, Reference Fraser, Holme, Martin, Whiteley, Montarello, Raw, Carver and Polkinghorne2019). Analysis of the mitochondrial cox1 gene of S. scabiei from vicuñas revealed a clade that was phylogenetically related to S. scabiei in canids (Fig. 3A).
On the other hand, several studies worldwide have analysed the ITS2 region of S. scabiei from different hosts (Zahler et al., Reference Zahler, Essig, Gothe and Rinder1999; Verdugo et al., Reference Verdugo, Espinoza, Moroni, Valderrama and Hernandez2016; Peltier et al., Reference Peltier, Brown, Ternent, Niedringhaus, Schuler, Bunting, Kirchgessner and Yabsley2017; Li et al., Reference Li, Sun, Xie, Zhou, Gu, Lai, Peng and Yang2018). In all of them, the results indicated a greater genetic variability, decreasing over the years, compared to the cox1 gene (Amer et al., Reference Amer, El Wahab, Metwaly Ael, Ye, Roellig, Feng and Xiao2014; Fraser et al., Reference Fraser, Charleston, Martin, Polkinghorne and Carver2016, Reference Fraser, Shao, Fountain-Jones, Charleston, Martin, Whiteley, Holme, Carver and Polkinghorne2017; Peltier et al., Reference Peltier, Brown, Ternent, Niedringhaus, Schuler, Bunting, Kirchgessner and Yabsley2017). In our study, the ITS2 region analysis of S. scabiei from vicuñas showed three known haplotypes, which have been previously identified from S. scabiei from Pyrenean chamois (Rupicapra pyrenaica) from Spain (Berrilli et al., Reference Berrilli, D'Amelio and Rossi2002), water buffalos (Bubalus bubalis) from Egypt (Amer et al., Reference Amer, El Wahab, Metwaly Ael, Ye, Roellig, Feng and Xiao2014), and humans from France and Japan (Fukuyama et al., Reference Fukuyama, Nishimura, Yotsumoto, Gushi, Tsuji, Kanekura and Matsuyama2010; Delaunay et al., Reference Delaunay, Herisse, Hasseine, Chiaverini, Tran, Mary, Del Giudice, Marty, Akhoundi and Hubiche2020), respectively. Likewise, five new haplotypes were identified in S. scabiei from vicuñas (Table 2). In addition, as other studies have demonstrated (Zahler et al., Reference Zahler, Essig, Gothe and Rinder1999; Gu and Yang, Reference Gu and Yang2008; Amer et al., Reference Amer, El Wahab, Metwaly Ael, Ye, Roellig, Feng and Xiao2014), the ITS2 nucleotide sequence polymorphism of S. scabiei from vicuñas was not associated with the host species and the geographic location (Fig. 3B).
As with other parasitic agents, host–parasite adaptation and geographic segregation play an essential role in understanding the epidemiology of this mite. For many years, it has been believed that the S. scabiei transmission could occur between host species and that their descent with new genetic variability could infect new animal species (Fain, Reference Fain1994; Fraser et al., Reference Fraser, Shao, Fountain-Jones, Charleston, Martin, Whiteley, Holme, Carver and Polkinghorne2017). In recent decades, molecular studies have attempted to better explain the epidemiology of S. scabiei in different host species (Amer et al., Reference Amer, El Wahab, Metwaly Ael, Ye, Roellig, Feng and Xiao2014; Fraser et al., Reference Fraser, Shao, Fountain-Jones, Charleston, Martin, Whiteley, Holme, Carver and Polkinghorne2017, Reference Fraser, Holme, Martin, Whiteley, Montarello, Raw, Carver and Polkinghorne2019; Peltier et al., Reference Peltier, Brown, Ternent, Niedringhaus, Schuler, Bunting, Kirchgessner and Yabsley2017; Bae et al., Reference Bae, Kim, Jung, Cha, Jeon, Lee, Kim, Chang and Kim2020). These studies demonstrated the existence of various genotypes analysing different molecular markers, as the cox1 gene, suggesting some host–parasite adaptation for S. scabiei, as supported by this study. Furthermore, the phylogenetic analysis of the S. scabiei cox1 gene indicates that canids are possibly involved in transmitting sarcoptic mange to vicuñas. However, to demonstrate this, it will be necessary to molecularly analyse S. scabiei from canids and other mammals from the Cuzco areas, adding the use of other genetic markers such as microsatellites.
The main limitations of our study are the small number of specimens analysed by each vicuña, the use of mites from the same geographic area, and not used S. scabiei from other host species, both domestic and wild. Although the analysis of cox1 showed an exclusive clade for vicuñas, with these limitations, we cannot conclude that S. scabiei mites from vicuñas or other South American camelids are exclusively adapted to these particular hosts. Therefore, it will be necessary to carry out future molecular studies such as population genetics and comparative genomics to help us determine the variety of strain, geographical segregation and the host–parasite adaptation for the S. scabiei from vicuñas from Peru. Likewise, this will help understand valuable and applicable concepts for the conservation of wildlife and its health, and their relationship with domestic animals and humans.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182021001931
Acknowledgements
The authors wish to thank Dr Henry Castelo Oviedo from the Sustainable Management of Vicuñas Project (Cuzco Regional Government) for the facilities provided during the samples collection.
Author contributions
J.I.P., J.M.A.T. and W.G. performed the animal evaluation and sample collection. L.A.G.P. conducted molecular analysis. L.A.G.P., H.C., M.T.L.U. and A.E.G. analysed the data. L.A.G.P. and H.C. wrote the manuscript. All authors read and approved the final manuscript.
Financial support
This research was supported by the Universidad Nacional Mayor de San Marcos – RR No 03202-R-18 and project number A18080611.
Conflict of interest
None.
Ethical standards
This study was reviewed and approved by the Forest and Wildlife Service of Peru (Servicio Nacional Forestal y de Fauna Silvestre – SERFOR) from Lima, Peru.







