INTRODUCTION
Schistosomiasis is a disease of profound medical and veterinary importance, infecting over 206 million people (Steinmann et al. Reference Steinmann, Keiser, Bos, Tanner and Utzinger2006). Among the species of schistosomes infecting humans, Schistosoma haematobium is responsible for the largest number of infections in sub-Saharan Africa with an estimated 112 million people infected with this species (van der Werf et al. Reference van der Werf, de Vlas, Brooker, Looman, Nagelkerke, Habbema and Engels2003), more than double the estimated figure for Schistosoma mansoni. However, S. haematobium, and in particular its genetic diversity, remains largely unstudied in comparison to S. mansoni, primarily due to the more demanding conditions for laboratory maintenance and a lack of available molecular markers for the former species. In the last 40 years, for example, there has been an almost 10-fold difference in the number of papers published on S. mansoni compared to S. haematobium (Rollinson, Reference Rollinson2009), and molecular studies are particularly lagging behind, especially since the recent publication of complete genome sequences for both S. mansoni and S. japonicum (Webster et al. Reference Webster, Oliveira, Rollinson and Gower2010; Zerlotini and Oliveira, Reference Zerlotini and Oliveira2010). However, as recently reviewed by Rollinson (Reference Rollinson2009), there are many important biological, clinical and epidemiological differences between uro-genital and intestinal schistosomiasis that argues that further targeted research on S. haematobium is a research priority.
Successes of recent disease control against S. haematobium at the national scale have been reported following mass drug administration (MDA) of affected communities with praziquantel (PZQ) with impressive reductions in helminth infection intensity, prevalence and associated morbidity e.g. (Fenwick et al. Reference Fenwick, Webster, Bosque-Oliva, Blair, Fleming, Zhang, Garba, Stothard, Gabrielli, Clements, Kabatereine, Toure, Dembele, Nyandindi, Mwansa and Koukounari2009; Stothard et al. Reference Stothard, French, Khamis, Basanez and Rollinson2009; Webster et al. Reference Webster, Koukounari, Lamberton, Stothard and Fenwick2009). The recent implementation of such large-scale MDA might be expected to lead to strong and prolonged evolutionary selection pressures on the parasites themselves, in particular for the evolution of drug resistance (Webster et al. Reference Gower, Shrivastava, Lamberton, Rollinson, Emery, Webster, Kabatereine and Webster2008). Our ability to monitor potential drug resistance in schistosomes, however, is hampered by a lack of molecular markers associated with PZQ action. Neutral markers, such as microsatellites, offer the opportunity to distinguish treatment failures (and thus putatively resistant parasites) from new infections when individual infected people are sampled over time, as well as to investigate microevolutionary forces, such as migration, gene flow and genetic drift, and to estimate effective population sizes and any changes mediated by mass treatment (Webster et al. Reference Gower, Shrivastava, Lamberton, Rollinson, Emery, Webster, Kabatereine and Webster2008). This should allow determination of the impact of treatment on the parasite population as a whole, and inform of likely refugia (van Wyk, Reference van Wyk2001) as well as the potential for the spread of alleles associated with drug resistance, and assess the genetic consequences of varying control strategies.
Mathematical models have been developed to study the potential evolution of drug resistance in lymphatic filariais, and have identified a number of characteristics, such as the extent of non-random mating and initial frequency of resistance alleles as important (Schwab et al. Reference Schwab, Churcher, Schwab, Basanez and Pritchard2006). However, evolution of drug resistance is only one dimension of potential helminth adaptation to human intervention through mass chemotherapy (Webster et al. Reference Gower, Shrivastava, Lamberton, Rollinson, Emery, Webster, Kabatereine and Webster2008). Large-scale use of chemotherapeutic agents is also likely to result in selection on parasite life-history traits, including potential trade-offs between survivorship, fecundity and/or transmission strategies (Davies et al. Reference Davies, Webster and Woolhouse2001), all traits that, in S. mansoni at least, have been shown to exhibit genetic variation and respond to laboratory selection (Gower and Webster, Reference Gower and Webster2004). Since egg production is the major cause of pathology in schistosome infections, change in reproductive strategies could impact on the intensity or morbidity of residual or untreated infections. Variations in parasite reproductive success or ‘costs’ linked with drug resistance are critical for its likely establishment and spread and probably account for the continued efficacy of PZQ against S. mansoni in Egyptian villages despite the isolation 10 years previously of parasites with decreased PZQ sensitivity (Botros et al. Reference Botros, Sayed, Amer, El-Ghannam, Bennett and Day2005). Moreover, as MDA may be predicted to impact on density-dependent regulation of parasite populations, there may be phenotypical variations in parasite reproduction with both implications for transmission and practical consequences for the measurement of drug and control programme efficacy. Estimating parasite reproductive success is difficult in natural infections, and in schistosomes in particular, since adult worm burdens cannot be measured directly; however, ‘sibship’ analyses of miracidial genotypes (whereby the number of families or ‘sibs’ are identified) might prove useful in assessing the reproductive success of individual schistosome genotypes, and any changes associated with treatment (Criscione et al. Reference Criscione, Poulin and Blouin2005). Genetic variation in reproductive success of individual genotypes is a pre-requisite for evolutionary selection to occur, but we do not yet have any empirical evidence to know if this applies in natural populations of schistosomes.
Molecular epidemiological studies may also be useful in interpreting prevailing patterns of infection. A feature of schistosome epidemiology is that infection intensity and disease severity vary considerably between geographical areas and between individuals within the same region (Chunge et al. Reference Chunge, Karumba, Ouma, Thiongo, Sturrock and Butterworth1995). Laboratory studies, albeit currently limited only to S. mansoni, have suggested that such variation in definitive and intermediate hosts may be attributed in part to differences in parasite strain (Nelson and Saoud, Reference Nelson and Saoud1968; Thiongo et al. Reference Thiongo, Madsen, Ouma, Andreassen and Christensen1997; Davies et al. Reference Davies, Webster and Woolhouse2001; Gower and Webster, Reference Gower and Webster2004), and there is preliminary evidence of an association of specific parasite genotypes with more intense pathology in S. haematobium (Brouwer et al. Reference Brouwer, Ndhlovu, Wagatsuma, Munatsi and Shiff2003). Thus if parasite genotype influences disease outcome, we might predict that isolates from children with a similar level of infection intensity, or clinical presentation, would be more closely related than would be expected by chance alone. Likewise, our prior laboratory work has demonstrated that infection of hosts by multiple strains or parasite genotype individuals may be associated with exacerbated host morbidity, as a direct consequence of intra-host parasite competition and more diverse infections may pose added burdens on the host immune system (Davies et al. Reference Davies, Fairbrother and Webster2002). Since damage from schistosome infections is so closely related to immune reactions to parasite eggs deposited in tissue, diversity of this infection may play an important role in development of pathology with heterogeneous and homogenous infections resulting in different clinical outcomes. Conversely, parasites may benefit from increased genetic diversity if this aids evasion of the immune response. Thus one may predict that genetic diversity per se may be important in terms of causing host morbidity as well as presence of any specific genotype(s).
Adult schistosomes cannot be directly sampled from living hosts due to their location in the blood vessels of the mesenteric system and traditionally schistosomes have been passaged through laboratory animals in order to obtain parasite material for analysis. Recent work has overcome the logistical and sampling bias problems associated with such passage and developed methods to directly genotype schistosome larvae collected from infected people (Shrivastava et al. Reference Shrivastava, Gower, Balolong, Wang, Qian and Webster2005; Sorensen et al. Reference Sorensen, Rodrigues, Oliveira, Romanha and Minchella2006; Gower et al. Reference Gower, Shrivastava, Lamberton, Rollinson, Emery, Webster, Kabatereine and Webster2007; Webster, Reference Webster2009), using microsatellite markers.
Here we report on the development of 16 novel polymorphic microsatellite markers for S. haematobium. Five of these markers, together with 3 previously published markers, were successfully combined in a multiplex PCR allowing genotyping of individual larval samples at up to 8 microsatellite loci. This assay was used to genotype the parasite populations of 47 children from 2 schools in the Ségou region of Mali sampled in February 2005, immediately prior to the introduction of a national schistosomiasis control programme. We aimed to characterize the population genetics of this untreated population, and identify potential associations of parasite diversity and/or genotype with infection intensity. We further aimed to use sibship analysis to quantify schistosome reproductive success in a natural population. These data will serve as a baseline against which to measure the effect of community-wide treatment with PZQ on parasite population genetics in this region, as well as providing a rare insight into the population genetics of S. haematobium in general. The tools developed will be useful for ongoing and future monitoring of MDA in other regions, as well as in Mali.
MATERIALS AND METHODS
Isolation of novel microsatellite loci
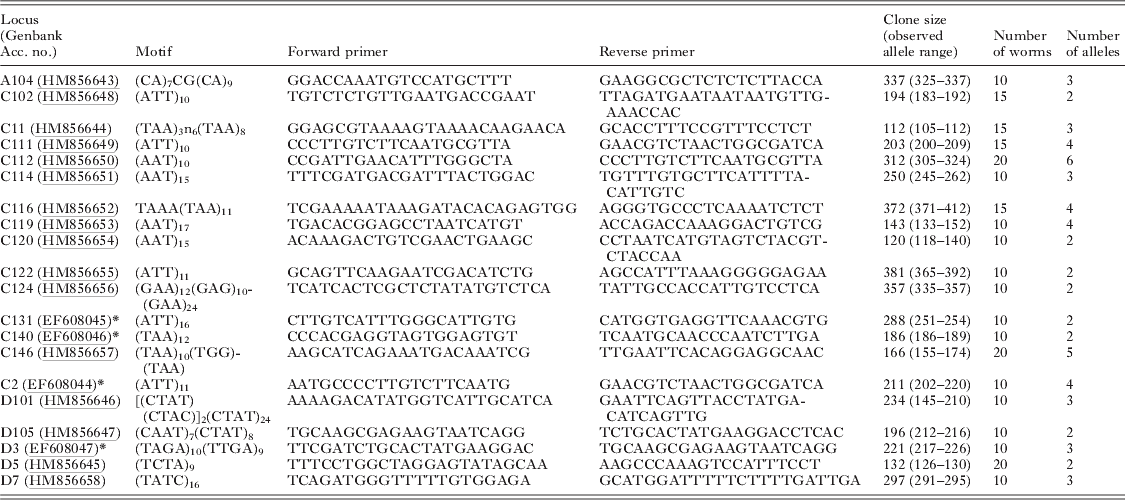
Four genomic libraries enriched for simple sequence repeats (CA, AAT, ATG, TAGA) were constructed using E. coli DH5 alpha and pUC19 vector by Genetic Identification Services (GIS, Chatsworth, CA, USA). DNA from adult S. haematobium worms originating from Mali, Tanzania, Zanzibar, Zambia, Uganda and Zimbabwe (held in the Liquid Nitrogen Collection of the Natural History Museum, London, originating from naturally infected snails and obtained after the first passage in laboratory animals) was pooled and used in library construction. Eighty seven microsatellite-containing clones were sequenced and PCR primers designed for 30 suitable novel sequences. Copies of the libraries for the sequencing of other potential microsatellite-containing clones are available from the corresponding author. Amplifiable loci were assessed for sensitivity, specificity and polymorphism using S. haematobium DNA from 10–20 individual adult worms of mixed geographical origin.
Genomic DNA was extracted from ethanol-preserved individual worms by digestion with proteinase K and transfer to Indicating FTA® Classic Cards (Whatman, Middlesex, UK). For DNA extraction, a 2·0 mm sample disk was removed from the Whatman FTA® cards using a Harris Micro Punch and incubated for 5 min in FTA® purification reagent (Whatman plc, Maidstone, Kent). The FTA® purification reagent was removed and replaced twice for a total of 3 washes. This was followed by 2×5 min incubations in TE buffer (pH 8·0). Samples were dried for 10 min at 56°C before use in PCR reactions.
PCR reactions were performed using a PCR-200 Thermal Cycler (MJ Research, UK). Amplifications were performed in 25μl reactions containing template ⩽1 μg DNA approximately, 0·05 μ m of each primer, 3 mm MgCl2, ultra-pure quality dNTP mix and HotStarTaq DNA Polymerase (Qiagen® Multiplex PCR kit, West Sussex, UK). Forward primers were fluorescently labelled using 6-FAM (Applied Biosystems, Cheshire, UK). All PCR set-ups were conducted in a PCR hood and used filter barrier pipette tips. Thermal cycling was performed with a step-down PCR beginning with an initial hot-start activation of 15 min at 95°C, followed by 40 cycles of 30 sec at 94°C, 90 sec at annealing temperature (2 cycles at each temperature from 60 to 51°C, followed by 20 cycles at 50°C), followed by a final extension at 60°C for 30 min. Products were diluted 5-fold in autoclaved deionized water and analysed using an ABI 3730 automated sequencer and Genescan®-LIZ500 size standard. All samples were run with positive and negative controls.
Development of a multiplex microsatellite assay for S. haematobium larvae and assessment of specificity and utility for other schistosome species
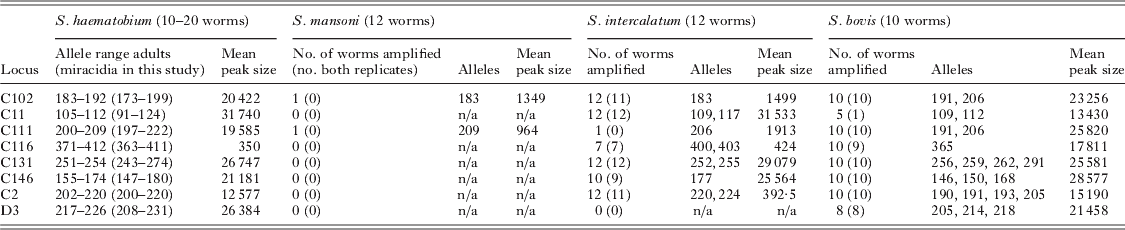
A multiplex PCR was developed suitable for use on miracidial samples collected on Whatman FTA® cards. Loci for use in the multiplex reaction were determined empirically based on the criterion of the maximal number of loci that could be reliably amplified in a single PCR reaction. Allele size range, strength and specificity of amplification and a lack of multiplex artefacts were the important inclusion factors. The final developed multiplex PCR assay (Table 2) used 5 novel and 3 previously published S. haematobium microsatellite loci (Golan et al. Reference Golan, Gower, Emery, Rollinson and Webster2007 and Table 1) using the reaction conditions described above. Novel primer pairs were designed for the previously published loci that were compatible with the multiplex reaction. Forward primers were fluorescently labelled using 6-FAM, VIC, NED and PET dyes (dye set – G5) (Applied Biosystems, Cheshire, UK), using different colours for alleles with overlapping size ranges (Table 2). A random selection of 20 adult schistosome worms was used to conduct both multiplex and single locus amplifications of each of the 8 loci used in the multiplex assays, under identical reaction and thermocycling conditions, in order to verify the robustness of the multiplex reaction in obtaining parasite genotypes. A further 12 adult worms were subject to 2 independent multiplex reactions in order to estimate genotyping error rates. The specificity of the assay and utility for other schistosome species was assessed by genotyping 12 S. guineensis (formerly termed S. intercalatum Kane et al. Reference Kane, Southgate, Rollinson, Littlewood, Lockyer, Pagès, Tchuem Tchuentè and Jourdane2003), 12 S. mansoni and 10 S. bovis worms from the Liquid Nitrogen Schistosome Collection of the Natural History Museum, London. Worms were derived after first passage in laboratory animals after collection of infected snails in Senegal (NHM342 for S. bovis) or São Tomé (NHM2657 for S. guineensis).
Table 1. Primer sequences and characteristics of 16 novel and 4 re-designed Schistosoma haematobium loci and amplification characteristics with 10–20 adult worms of mixed geographical origin
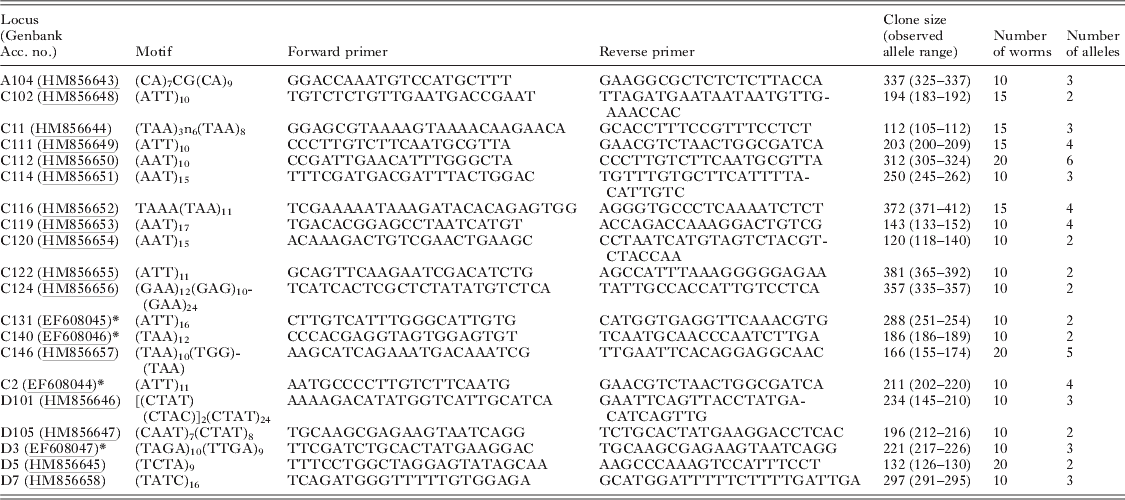
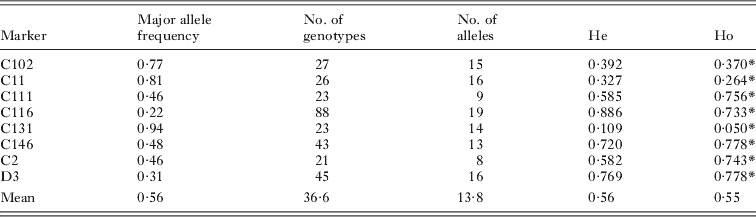
Table 2. Analysis of 862 larval schistosomes (miracidia) from Malian school children with a novel multiplex PCR for genotyping at 8 microsatellite loci
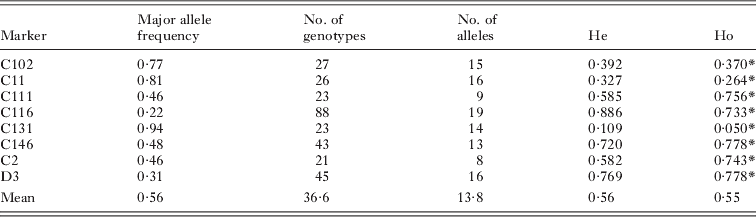
* Significant deviation from Hardy-Weinberg equilibrium.
Population genetic analysis of S. haematobium from Malian schoolchildren
Urine samples positive for S. haematobium group eggs were collected from 22 children (9 boys and 13 girls) at Bambougou-Wèrè School (13°38′0″N, 6°5′0″W) and from 25 children (16 boys and 9 girls) at Dougouba School (13°34′33″N, 6°06′44″W) in the Ségou region of Mali in February 2005, prior to the introduction of a National Schistosomiasis Control Program (see Fig. 1 for location of parasite collections). Ethical approval was obtained from the Imperial College Research Ethics Committee (ICREC), Imperial College London, UK in combination with the ongoing Schistosomiasis Control Initiative activities. Within Mali, official ethical clearance was not required because all aspects of Monitoring and Evaluation were carried out in the framework of the disease-control activities implemented and approved by the Ministry of Health (MOH). Before conducting the study, the MOH-approved plan of action had been presented and adopted by regional and local administrative and health authorities. Informed oral consent was received from the community leaders (via community forum), teachers, parents, and teachers’ directors, as is appropriate in many parts of Mali given the social underpinnings. Mean prevalence of S. haematobium at a baseline survey conducted in Mali at the setting up of the National Control Program, including the Ségou region (but unfortunately not including the schools from which parasite samples were collected) was 59% with an arithmetic mean infection intensity of 43 eggs/10 ml of urine, and an overall correlation between infection intensity and pathology as detected by ultrasound examination (Koukounari et al. Reference Koukounari, Sacko, Keita, Gabrielli and Landoure2006). Children in this study were randomly selected from children identified to be infected with S. haematobium by urine filtration and asked to provide a further urine sample for the isolation of S. haematobium parasites. Child age, sex and the intensity of the infection (as measured as the arithmetic mean number of eggs passed by 10 ml of urine) were recorded for each child. All infected children were treated with PZQ at 40 mg/kg.

Fig. 1. Map showing location of study in Ségou region of Mali. Parasite samples were collected from 2 schools, Bambougou-Wèrè, and Dougouba, situated close to the River Niger. The arrow indicates the direction of the river flow.
Miracidia were hatched from eggs from urine samples using bottled spring water. For DNA storage, following hatching of eggs, individual miracidia were picked up using a glass pipette under a binocular microscope and transferred to Whatman FTA® indicator cards (Whatman plc, Maidstone, UK) in a volume of 5 μl of bottled spring water and allowed to dry for 1 h. Addition of the sample activates chemicals in the cards that lyse cells, inactivate proteins and immobilize the genomic nucleic acids. This allows DNA samples to be stored and transported at room temperature. Up to 40 miracidia were collected per child, depending on infection intensity, with a mean sample size of 20·6 miracidia per child. Individual miracidia were subject to DNA extraction and multiplex PCR for 8 microsatellite loci as described above.
Data analysis
Diversity of infections
Allele sizes were calculated in Genemapper v. 4 and manually checked. Genetic data analysis (GDA) version 1.1 (Lewis and Zaykin, Reference Lewis and Zaykin2001) was used to calculate the total number of alleles per locus, the number of private alleles per locus (i.e. alleles exclusive to a single population), individual allele frequencies, and the expected and observed heterozygosity for the parasite population of each child. Since the total number of alleles is affected by sample size, which varied between individual children due to differences in infection intensity, allelic richness – a measure of allelic diversity which rarifies to the smallest sample size and hence is independent of sample size – was calculated in FSTAT vs 2.9.3.2(Goudet, Reference Goudet2002).
Hardy-Weinberg equilibrium analyses, testing the hypothesis that observed diploid genotypes are the product of random union of gametes (i.e. that the population is randomly mating), over the whole population and within the populations of individual children were carried out using Arlequin 2.000 (Schneider et al. Reference Schneider, Roesli and Excoffier2000). To detect significant departure from this equilibrium, a modified version of the Markov-chain random walk algorithm was used, a test analogous to Fisher's exact test using a contingency table of observed allele frequencies and the number of alleles through 100 000 permutations (Guo and Thompson, Reference Guo and Thompson1992). Genotypic disequilibrium tests for pairs of loci overall and within populations were carried out using Arlequin. Bonferroni corrected p values for multiple comparisons within each sample were used.
Differences in the mean number of loci per child, allelic richness and heterozygosity deficiency (He-Ho) were investigated using general linear modelling (Minitab, Minitab Inc, State College, PA), controlling for the number of miracidia sampled. School, age, gender and their interactions are presented as independent variables. A non-parametric permutation test, with 15 000 permutations, was also used to investigate any difference in the allelic richness of parasite populations between the two schools (FSTAT vs 2.9.3.2).
Population structure
As a measure of genetic distance between the parasite populations of individual children, a matrix of Cavalli-Sforza and Edwards’ chord distances (Cavalli-Sforza and Edwards, Reference Cavalli-Sforza and Edwards1967) was estimated using POPULATIONS 1.2.28 (Langella, Reference Langella1999) and visualized using a Neighbour-joining clustering algorithm (NJ). The reliability of phenograms was assessed by bootstrapping over loci with 10 000 replications.
Hierarchical Wrights Fst statistics measuring evidence of genetic differentiation between schools and between children within schools were calculated in an analysis of molecular variance (AMOVA) procedure in Arlequin 2.000. P-values were calculated by 100 000 random permutations. A hierarchical analysis was necessary as it accounts for the potential relatedness of miracidia within individual children, which will tend to inflate estimates of classical Wrights Fst statistics (Rudge et al. Reference Rudge, Carabin, Balolong, Tallo, Shrivastava, Lu, Basanez, Olveda, McGarvey and Webster2008).
Sibship analyses
The estimation of full-sib and half-sib relationships between all miracidia was carried out using Colony (V2.1) (Wang, Reference Wang2004). All sampled miracidia were partitioned simultaneously into genetic groups of variable size, corresponding to inferred familial groups. This program implements a maximum likelihood algorithm when comparing different sibship configurations and also allows for genotyping error (Wang and Whitlock, Reference Wang and Whitlock2003). Multiple runs were conducted to ensure convergence of maximum-likelihood solutions. Sibship data were used to determine the presence of related miracidia between individual children and schools, the size of sibship clusters and estimate the number of family groups/unique adult worm genotypes present in individual children. The reproductive success of each parasite genotype within individual children was estimated as the number of miracidia per adult female genotype. Inferred adult worm genotypes were not used directly in analyses and it should be noted that due to the life cycle of schistosomes, whereby clonal amplification takes place in the intermediate snail host immediately prior to human infection, the number of parasite genotypes present may not reflect the actual number of worms present in the human host, but is instead a measure of the reproductive success of the original snail-infecting parasite.
Infection intensity and associations with population genetic data
Differences in infection intensity between children were investigated by general linear modelling (Minitab, Minitab Inc, State College, PA) using school, child gender and child age and their interactions as dependent variables. Associations between infection intensity and measures of allelic diversity were also investigated using general linear modelling (Minitab, Minitab Inc, USA) using allelic richness and observed heterozygosity as independent variables and intensity of infection as a categorical variable with infections classified as very heavy (>400 eggs/10 ml), heavy (100–399 eggs/10 ml) and light (<99 eggs/10 ml), controlling for age, school and gender. A non-parametric permutation test, with 15 000 permutations, was also used to investigate a difference in the allelic richness of parasite populations between very heavy (>400 eggs/10 ml), heavy (100–399 eggs/10 ml) and light (<99 eggs/10 ml) infections both within and across the two schools (FSTAT vs 2.9.3.2). Differences in the number of unique parental genotypes and number of familial clusters present in individual children were investigated using general linear modelling (Minitab, Minitab Inc, State College, PA), using school, child age and child gender and their interactions, and infection intensity [very heavy (>400 eggs/10 ml), heavy (100–399 eggs/10 ml) and light (<99 eggs/10 ml)] as the dependent variables.
RESULTS
Isolation of novel microsatellite loci
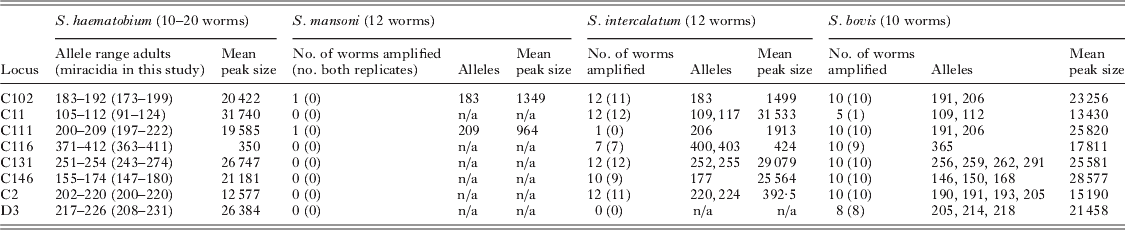
Sixteen novel polymorphic loci (1 dinucleotide, 11 trinucleotides and 4 tetranucleotides) were isolated for S. haematobium (Table 1). They comprised 9 perfect repeats, 3 compound repeats and 4 imperfect repeats with a repeat size ranging from 9 to 46. Five of these loci were successfully combined with re-designed primers for 3 previously published loci (Golan et al. Reference Golan, Gower, Emery, Rollinson and Webster2007) (Table 2) to create a multiplex assay capable of genotyping individual field-collected S. haematobium miracidia at 8 microsatellite loci in a single PCR reaction. For the 20 adult worms subject to both single locus and multiplex amplification there was 99% agreement between the reactions. Of the 12 worms subject to repeat genotyping, 45 alleles were identical in the independent repeats, and 3 discrepancies were identified, giving an overall low mean error rate of 0·85%. No errors were detected in loci C11, C111, C116, C113, C146 and C2, while there was a single error in loci C102 and 2 errors in loci D3 (both in the same individual). These were likely due to scoring errors, since traces were similar. These loci were not amplifiable in S. mansoni, but were strongly cross-reactive and produced similar allelic sizes with related members of the S. haematobium group including S. bovis and S. guineensis (Table 3), although peak heights for a fixed volume of PCR product was lower for some loci in the latter species.
Table 3. Specificity and utility of multiplex microsatellite assay for other schistosome species, including number of worms amplified (number of worms where both replicates amplified), allele sizes and mean microsatellite peak size for fixed PCR product volume loaded
Population genetic analysis of S. haematobium group parasites from Malian schoolchildren
Analysis was restricted to miracidia with at least 4 allele calls. A total of 968 miracidia from FTA cards were subject to multiplex PCR and a total of 862 were analysed, resulting in an amplification success of 89·0%. Failure to amplify most likely results from the absence of template DNA (failure to capture a miracidium is relatively easy during collection) and/or poor quality DNA template. Although there was no evidence of the larger S. bovis eggs in our urine samples, nor reported evidence of hybrids in this region of Mali, since S. haematobium/S. bovis hybrids have recently been identified in other parts of West Africa (Huyse et al. Reference Huyse, Webster, Gelhof, Stothard, Diaw, Polman and Rollinson2009), and would also not be distinguishable with our loci, we will refer to the isolated parasites as ‘S. haematobium group’ in order to be strictly accurate.
Infection intensity
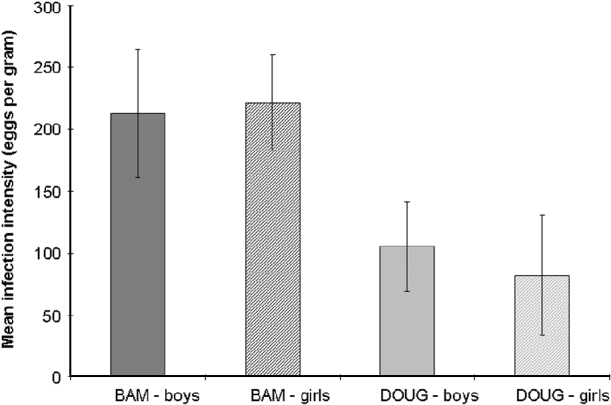
The mean age of children at Bambougou-Wèrè School was significantly lower than at Dougouba School (means 9·04 years compared to 11·52 years F1, 45=14·6, P<0·001). There was a significant difference in the intensity of the children's infections between the two schools (F1, 43=7·75, P=0·008; Fig. 2), with a higher mean intensity at Bambougou-Wèrè School and this was associated with a reduction in the intensity of infection with age. There was no difference in infection intensity between boys and girls at either of the schools.
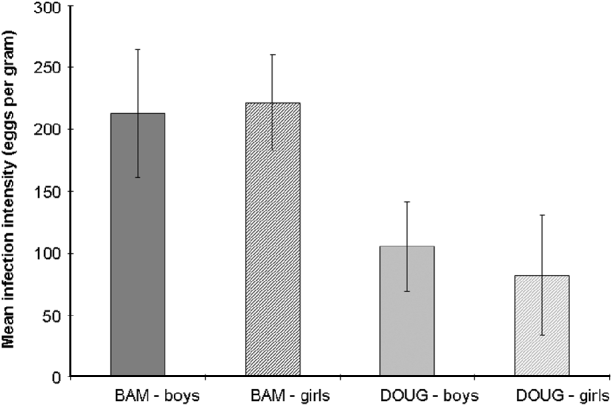
Fig. 2. Mean Schistosoma haematobium infection intensity (eggs/10 ml) of 23 boys (solid) and 22 girls (hatched) from 2 schools (Bambougou-Wèrè – BAM; mean age 9·04 years) and (Dougouba – DOUG; mean age 11·52 years) in the Ségou region of Mali.
Diversity of infections
Allelic diversity
All loci were highly polymorphic and the number of alleles per locus in the whole dataset ranged from 8 to 19 (Table 2). There was considerable variation between individual children in the diversity of their larval parasite populations, as shown by variations in the allelic richness of sampled populations (Fig. 3). However, there was no difference in the mean allelic richness between the parasites of children of different schools (F1, 42=0·42, P=0·52), genders (F1, 42=1·14, P=0·29) or ages (F1, 42=0·20, P=0·66) as assessed by general linear modelling. This result was supported by a non-parametric permutation test which found no evidence of a difference in allelic richness between the two schools (P=0·84). As expected, the total number of alleles was affected by miracidial sample size (F1, 42=41·87, P<0·001), but there was no further difference in the mean total number of alleles in larval parasite populations of children of different schools (F1, 42=0·41, P=0·53), genders (F1, 42=1·78, P=0·19) or ages (F1, 42=1·26, P=0·27) (Fig. 4a and b).
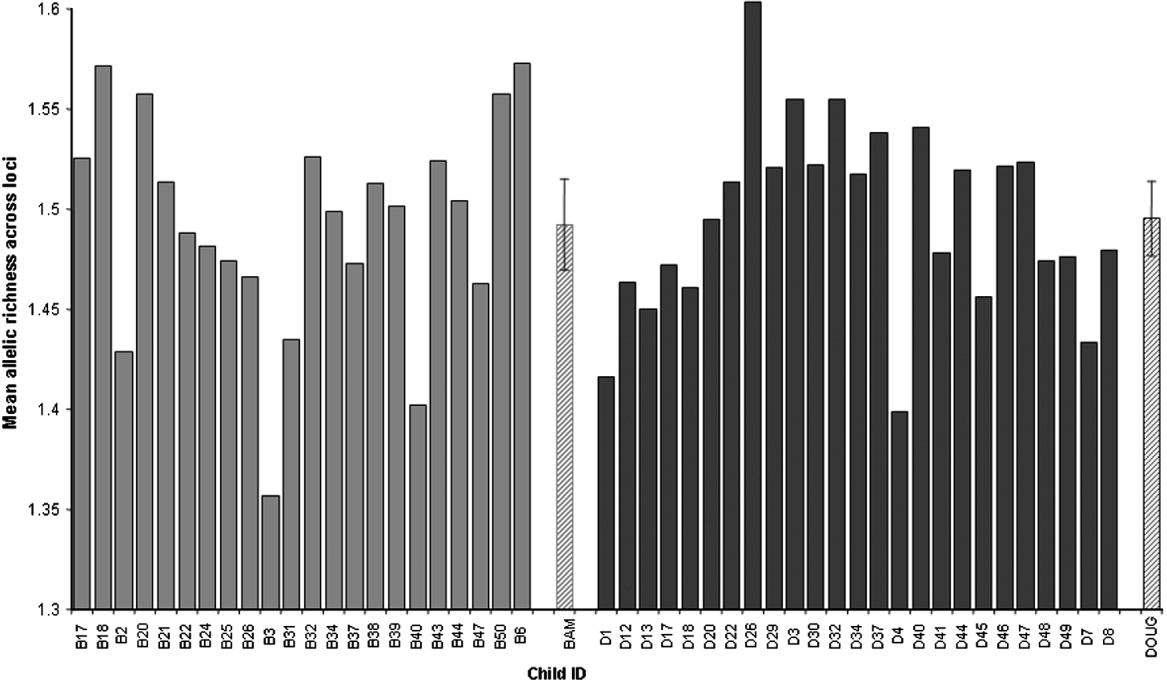
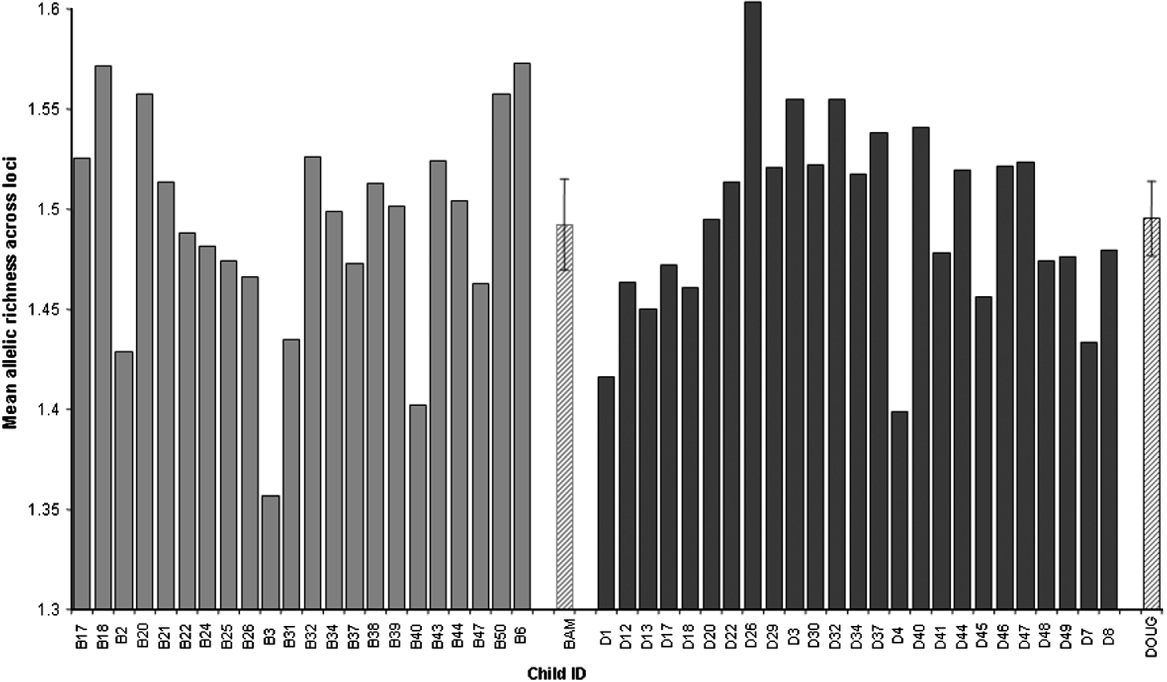
Fig. 3. Mean allelic richness of the Schistosoma haematobium larval populations of individual children in two schools (Bambougou-Wèrè – B) and (Dougouba – D) and the mean allelic richness (±95% c.i.) for each school (Bambougou-Wèrè – BAM) and (Dougouba – DOUG) (hatched area).
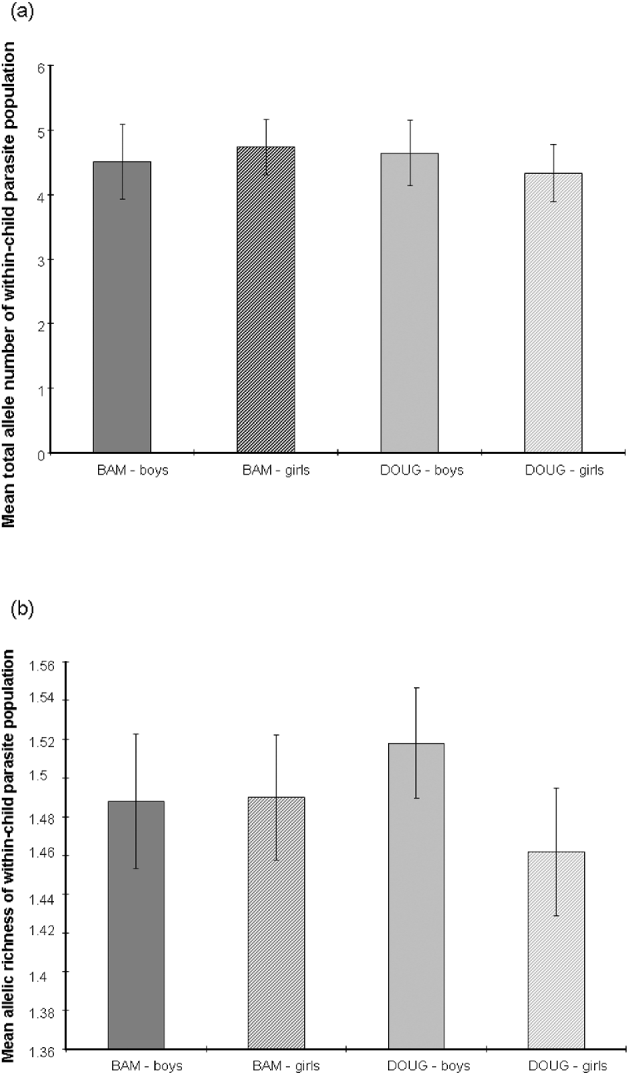
Fig. 4. Mean (a) total number of alleles (±95% c.i.) and (b) allelic richness (±95% c.i.) of the Schistosoma haematobium larval populations of individual boys (solid) or girls (hatched) in 2 schools (Bambougou-Wèrè – BAM) and (Dougouba – DOUG).
Major allele frequency(s) were high (Table 2), and 73% of alleles were only present at low frequency (<0·05%). Considering the whole dataset, there were 21 private alleles (i.e. only found within the parasite population of 1 child), found in 15 children. Within schools, there were 16 private alleles in Bambougou-Wèrè School (found in 13 children) and 16 alleles (in 11 children) in Dougouba School.
Heterozygosity
The expected and observed heterozygosity averaged across the 8 loci of the larval parasite populations of the 25 children from Bambougou-Wèrè School and 22 children from Dougouba School are shown in Fig. 5a and b respectively. There were individual children at each school that showed both an excess and a deficit of heterozygotes but there was no overall difference in mean expected versus observed heterozygosity at either school, or in mean difference in expected and observed heterozygosity between children of different schools (F1, 42=0·46, P=0·503). However, overall parasites from boys had relatively higher levels of inbreeding (mean Ho-He=−0·0088±0·0143) than parasites recovered from girls (mean Ho-He=0·038±0·0148) (F1, 42=4·79, P=0·03).

Fig. 5. Mean (across loci) expected (He) and observed heterozygosity (Ho) for Schistosoma haematobium larval populations of individual children (solid) and averaged over the school (hatched) from (a) Bambougou-Wèrè and (b) Dougouba school, Ségou region, Mali.
All individual loci showed evidence of Hardy-Weinberg (HW) disequilibrium when considered across the whole population (Table 2). However, within individual children, the vast majority of loci were in HW equilibrium with an average of 0·8 loci per child in disequilibrium (range 0–3), with no consistent pattern between children in affected loci. Similarly there was evidence of linkage disequilibrium across the dataset as a whole, although little evidence within the populations of individual children.
Associations of diversity with infection intensity
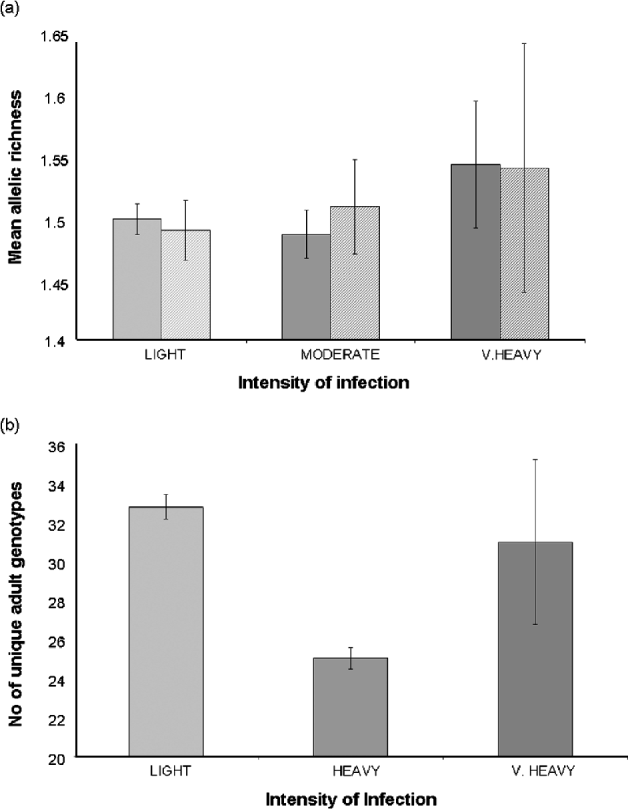
There was no significant difference in the mean allelic richness of the larval parasite populations of children with very heavy (>400 eggs/10 ml), heavy (100–399 eggs/10 ml) or light (<99 eggs/10 ml) infections (F2, 43=1·41, P=0·255) (Fig. 7a) nor in the number of miracidia sampled (F2, 44=1·06, P=0·39) nor the total number of alleles (F2, 44=1·10, P=0·342) with children of different infection intensities. This was supported by a non-significant permutation test of a difference in the allelic richness of parasite populations between very heavy, heavy and light infections either across the two schools (P=0·13) or within schools (Bambougou-Wèrè, P=0·28; Dougouba, P=0·10). There was a significant positive association between high infection intensity and the number of private alleles, particularly due to a high frequency of private alleles in the heaviest intensities of infection (F1, 45=5·05, P=0·03; Fig. 7b).
Population structure
A Neighbour-Joining (NJ) phenogram based on Cavalli-Sforza and Edwards' chord distances between the parasite populations of individual children is shown in Fig. 6. There was some evidence of clusters of parasite populations between children of the same school, but no evidence of a substantial separation between the schools. Similarly, there were no clear patterns associated with gender, age or infection intensity. Bootstrap support was low, indicative of low levels of population genetic variation between the parasite populations of individual children. This was confirmed by the analysis of molecular variance analysis (Table 4). There was very low yet significant genetic differentiation between the parasites collected in the two schools, Fst=0·004, P=0·0004) and between parasites from individual schools (Fst=0·001, P=0·002). However, as shown in Table 4, this represented only 0·35% and 0·1% of the total variance respectively, with the remaining 99·5% of the total variance attributable to variation within the parasite populations of individual children.
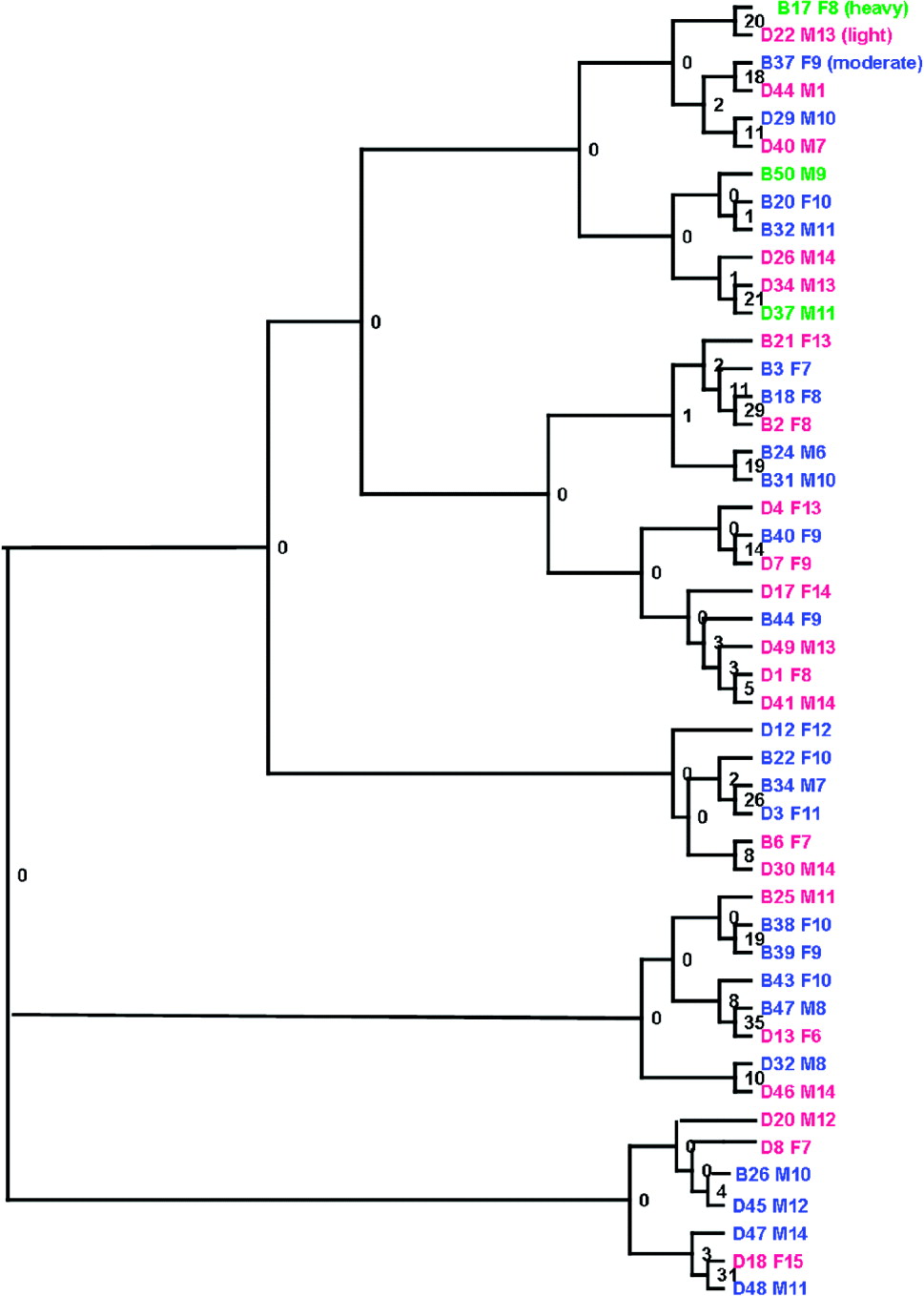
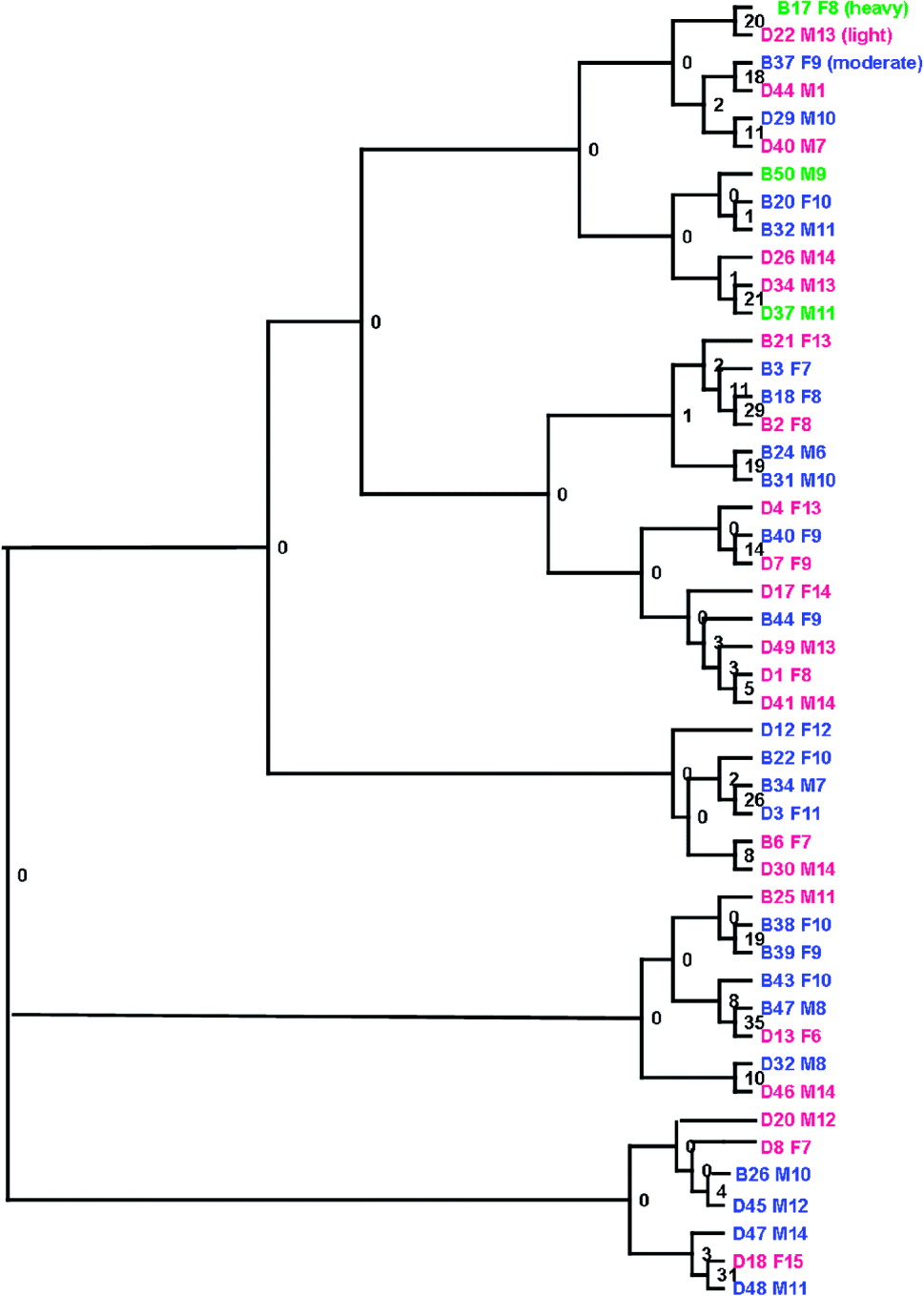
Fig. 6. Neighbour-joining clustering of Schistosoma haematobium larval populations from children with heavy (>400 eggs/10 ml) [green], light (<99 eggs/10 ml) [pink], moderate (100–399 eggs/10 ml)[blue] infection intensity. Branches represent infrapopulations of individual children identified by school – [B (Bambougou-Wèrè), D (Dougouba)] and child ID followed by sex (M – male or F – female) and child age in years.
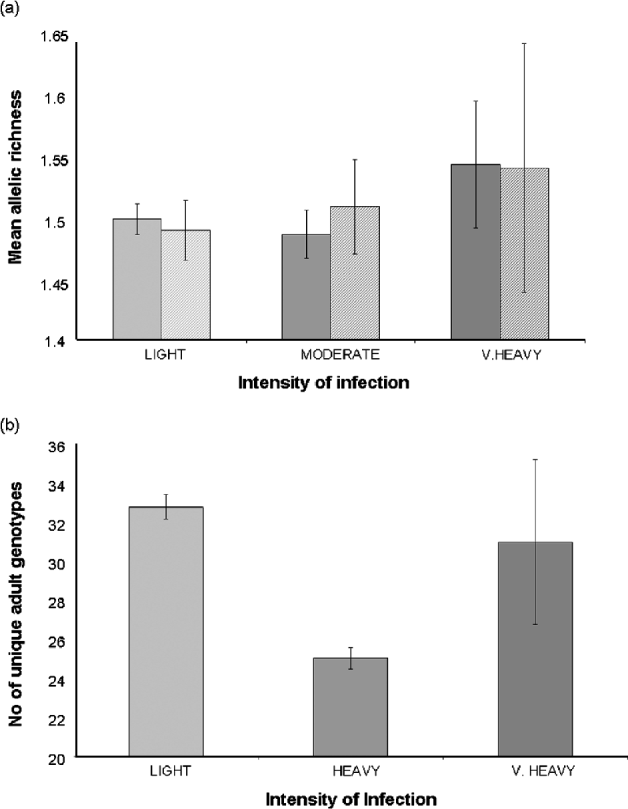
Fig. 7. (a) Mean allelic richness of larval Schistosoma haematobium parasite populations; (b) Number of private alleles; (c) Number of unique adult genotypes inferred from children with light (<99 eggs/10 ml), heavy (100–399 eggs/10 ml) or very heavy infections (>400 eggs/10 ml).
Table 4. Analysis of molecular variance showing the amount of variation between schools, within schools and within individual children within schools

Sibship analysis
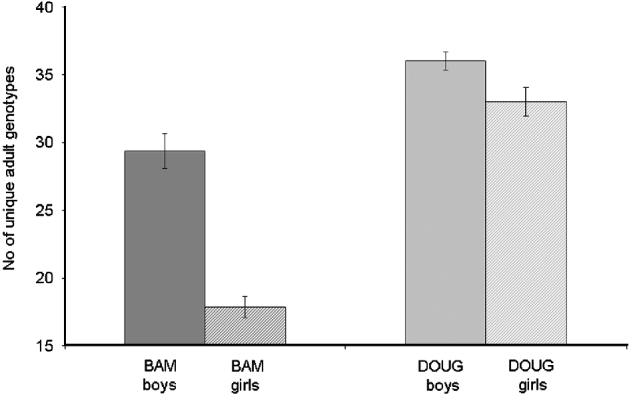
There were 67 familial clusters (full- and half-sibships) identified within the dataset, which ranged in size from 2 to 111 miracidia (mean 12·8). Fifty nine (88%) clusters were present in miracidia collected from both of the two schools, with those exclusive to a single school being small in size (mean=4·0; range=2–7). There was no overall association between the number of unique parental genotypes inferred and the intensity of infection (F1, 44=2·05, P=0·14). However, when we excluded the 3 children with the heaviest infections from the analysis, we found that counter-intuitively children with light infections (<99 eggs/10 ml) had more diverse adult worm infections than those with high infections (100–400 eggs/10 ml) (F1, 42=4·06, P=0·05) (Fig. 7c). The number of unique parental genotypes inferred per child was significantly higher in children from Dougouba school (mean=33·8) compared to those from Bambougou-Wèrè school (mean=23·1; F1, 43=7·39, P=0·009) and significantly lower in girls (mean 24·8) than boys (mean=32·0, F1, 43=4·31, P=0·04) (Fig. 8). There was no further association with child age (F1, 43=0·00, P=0·98), although it is noted that the average age of children was significantly higher at Dougouba school. Similarly, the number of familial clusters was significantly higher in Dougouba (F1, 43=6·97, P=0·012) and in boys (F1, 43=6·40, P=0·015). Within schools, however, there was no association between the number of unique parental genotypes inferred (Bambougou-Wèrè: F1, 19=0·04, P=0·85 Dougouba: F1, 22=0·17, P=0·69) or the number of full sibship clusters (Bambougou-Wèrè: F1, 19=0·69, P=0·42; Dougouba F1, 22=0·18, P=0·68) and child age. At Bambougou-Wèrè school, both were significantly increased in boys (number of parental genotypes; F1, 19=6·28, P=0·02; number of full sibship clusters F1, 19=9·03, P=0·007). Mean number of parental genotypes and number of full sibship clusters were also higher in boys at Dougouba school, but this was not a significant difference (number of parental genotypes: F1, 22=3·38, P=0·08; number of sibship clusters: F1, 22=0·49, P=0·48) (Fig. 8). Over the whole dataset, the number of miracidia per familial cluster was negatively associated with age (F1, 43=7·57, P=0·013) and was lower in boys than girls (F1, 43=4·58, P=0·046) – in other words, older children and boys had a reduced number of miracidia per sibship cluster.
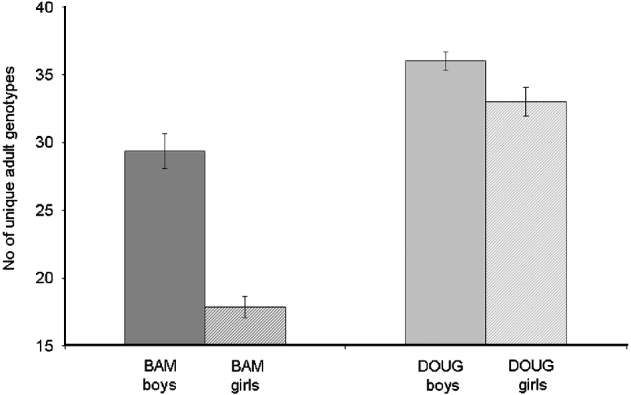
Fig. 8. Mean number of (a) unique adult worm genotypes and (b) number of full-sib clusters found in individual children from boys (solid) and girls (hatched) from 2 schools (Bambougou-Wèrè – BAM) and (Dougouba – DOUG) in the Ségou region of Mali.
DISCUSSION
Molecular epidemiological studies of schistosomes provide opportunities to investigate many important topics, such as the contribution of parasite genetics to variation in disease burden and pathology (Brouwer et al. Reference Brouwer, Ndhlovu, Wagatsuma, Munatsi and Shiff2003), the genetic consequences of various control activities for the parasite population (Curtis et al. Reference Curtis, Sorensen and Minchella2002; Norton et al. Reference Norton, Gower, Lamberton, Webster, Lwambo, Blair, Fenwick and Webster2010), patterns of recruitment and transmission in endemic areas (Sire et al. Reference Sire, Durand and Pointier1999) the potential for the circulation of strains differentially adapted to human and reservoir hosts (Rudge et al. Reference Rudge, Carabin, Balolong, Tallo, Shrivastava, Lu, Basanez, Olveda, McGarvey and Webster2008; Lu et al. Reference Lu, Rudge, Wang, Donnelly, Fang and Webster2010) and the likely evolution and spread of drug resistance (Webster et al. Reference Gower, Shrivastava, Lamberton, Rollinson, Emery, Webster, Kabatereine and Webster2008). We report here the development of 16 novel microsatellite loci for study of the major human pathogen, S. haematobium, and the development of the first multiplex assay capable of genotyping individual S. haematobium larval miracidia at 8 microsatellite loci, thus providing tools for the field collection, room temperature DNA storage and robust genetic characterization of this highly neglected parasite. DNA samples can be collected with minimal equipment in potentially remote village locations and may be stored at room temperature for several years, facilitating transfer to central archives or collaborators, which may be particularly pertinent for the temporal record of the effects of mass chemotherapy on parasite populations, and allows easy storage of samples should, for example, molecular markers associated with PZQ resistance be developed. Whole genome amplification (WGA) reactions are currently being standardized to overcome the limitation of this methodology which is that sufficient DNA is usually only obtained for a single PCR reaction (Gouvras, Reference Gouvras2010).
We further report that this microsatellite assay was not cross-reactive with S. mansoni, which are sometimes found in urine samples due to very high S. mansoni infection intensities, contamination during sample collections or most likely due to heterologous pairings between S. haematobium males and S. mansoni females (Cunin et al. Reference Cunin, Tchuem Tchuente, Poste, Djibrilla and Martin2003), in which S. mansoni eggs are produced parthogenetically in the preferred oviposition site of the male S. haematobium worms. The amplifications in single worms for 2 of the markers would not be scored under our scoring procedure, where individuals with fewer than 4 alleles scored are discarded. However, this assay can be used to successfully genotype other members of the S. haematobium group including S. guineensis (formerly termed S. intercalatum; Kane et al. Reference Kane, Southgate, Rollinson, Littlewood, Lockyer, Pagès, Tchuem Tchuentè and Jourdane2003) and the cattle schistosome S. bovis. This is perhaps not unsurprising considering the close genetic relationships of these species. Although this means that it is not possible to identify the schistosome species present within the S. haematobium group, by use of our microsatellite assay, we believe it is essential for robust population genetic studies given the existence of natural hybridization within the S. haematobium group (Huyse et al. 2009) since otherwise any hybrids present would be erroneously scored as homozygotes (since only the haematobium-derived genetic material would be genotyped). Moreover stable S. bovis – S. haematobium hybrids have recently been documented in Senegal (Huyse et al. 2009) resulting from bidirectional introgressive hybridization, and thus these populations are in effect acting as one large population from a genetic perspective and thus should usefully be considered together in population genetic studies. In contrast, in S. haematobium – S. mansoni pairings, though these infections have very different morbidity and epidemiological dynamics relative to single species infections (Koukounari et al. Reference Koukounari, Donnelly, Sacko, Keita, Landoure, Dembele, Bosque-Oliva, Gabrielli, Gouvras, Traore, Fenwick and Webster2010), it would not be expected that genetic material was being exchanged between populations. Though there was no evidence from egg morphologies for the presence of other schistosome species or hybrids in this study in Mali, the presence of any S. haematobium group hybrids may not have been detected using the molecular genotyping used and unfortunately there was no further material to allow mitochondrial bar-coding. Although these parasites may thus be better described as S. haematobium group parasites, we do not, however, feel that the presence of hybrids would alter the conclusions of our study regarding the presence of population structure, associations of genetic diversity with age, sex and infection intensity or utility of microsatellite markers for monitoring schistosomiasis control. We would recommend that future population genetic studies, now that WGA methodologies are available to increase the genetic material available (Gouvras, Reference Gouvras2010), include both mitochondrial barcoding to identify schistosome species – with important epidemiological ramifications for example, for the presence of reservoir hosts and snail species acting as intermediate hosts (Huyse et al. 2009) – and more polymorphic nuclear markers such as these microsatellite loci for more general population genetic analyses and monitoring of drug treatment success.
We used both traditional population genetic methodology of larval samples and maximum likelihood inference of familial relationships in order to investigate patterns of genetic variability in the parasite populations of 47 children from 2 schools in the Ségou region of Mali, immediately prior to the introduction of a national MDA control programme for schistosomiasis. This is the first report of the population genetics of S. haematobium group larvae using robust and variable co-dominant markers. Overall we demonstrated that there was considerable variability in these parasite populations with the mean number of alleles reported of a similar magnitude to those reported for S. mansoni (Agola et al. Reference Agola, Steinauer, Mburu, Mungai, Mwangi, Magoma, Loker and Mkoji2009; Norton et al. Reference Norton, Gower, Lamberton, Webster, Lwambo, Blair, Fenwick and Webster2010) and S. japonicum (Rudge et al. Reference Rudge, Carabin, Balolong, Tallo, Shrivastava, Lu, Basanez, Olveda, McGarvey and Webster2008). Similarly, we found only limited evidence of genetic differentiation between these untreated S. haematobium group populations, as evidenced by significant, yet very small Wrights Fst, statistics between the two schools, and between the parasite populations of individual children. The majority of the variation was found within individual children. This was supported by analyses of heterozygosity levels, which showed an overall deviation from H-W equilibrium across the entire dataset, although much less support for H-W disequilibrium within individual children (though see later discussion). This could be explained by non-random mating of parasites across the children, and, for the most part, random mating within children (Wahlund effect). This is not surprising given the life cycle of schistosomiasis, where sexual reproduction takes place within individual hosts. Substantial gene flow in this parasite population was supported by the high frequency of dominant alleles, a lack of clustering of parasite populations from the same school and, using sibship analysis, by the presence of the same familial clusters in multiple children and schools (Lu et al. Reference Lu, Rudge, Wang, Donnelly, Fang and Webster2010). Thus it suggests that a small amount of genetic subdivision occurs due to the structuring of parasite populations within individual hosts, but substantial gene flow has counteracted any further differentiation between individual children and the two geographical areas. Clustering analyses found little evidence of genetic similarity between children of different ages, genders or infection intensities. This study was conducted in a small geographical area, in 2 villages approximately 8 kilometres apart, and directly connected by the River Niger. Thus it is perhaps not surprising that high levels of gene flow exist, due to the movement of both infected people and potential dispersal of infected snails. Furthermore children might be expected to sample a number of transmission sites (Agola et al. Reference Agola, Steinauer, Mburu, Mungai, Mwangi, Magoma, Loker and Mkoji2009), thus facilitating gene flow. The effect of population subdivision and gene flow on the evolution of drug resistance is complex since low levels of genetic substructure may decrease the rate of development of drug resistance as it will reduce the frequency of resistant alleles coming into contact with each other, assuming resistance develops as a recessive trait, whilst conversely high gene flow will tend to spread rare traits, such as emerging resistance, to neighbouring populations. Further studies are necessary to determine the extent of population structure over larger geographical scales, and any changes associated with drug treatment.
Microsatellite analysis is particularly powerful since it allows investigation of the parasite populations of individual hosts. Over-dispersion is a characteristic of helminth infections, and has many consequences both with regard to their population biology and to public health since heavily infected individuals are simultaneously at highest risk of morbidity and are the major source (up to 80%) of environmental contamination. We investigated the variability of parasite populations in relation to characteristics of individual children, such as age, sex and the intensity of parasite infection. We first assessed the genetic diversity of the larval parasite populations, and concluded there was considerable variation between the parasite populations of individual children. This variation was not, however, associated with the intensity of infection, as might be expected if diverse infections were more or less costly to the host. Similarly there was no difference in the mean diversity of infections in the two schools, despite significantly higher mean infection intensity at Bambougou-Wèrè. If parasite genetics play a direct role in pathogenesis, we would expect closely related parasites to be associated with similar disease progression (Ferreira et al. Reference Ferreira, Nair, Hyunh, Kawamoto and Anderson2002). Clustering analysis, however, found little evidence of similarity of the parasite populations of children of differing ages, genders or infection intensities. This might suggest that host (King et al. Reference King, Blanton, Muchiri, Ouma, Kariuki, Mungai, Magak, Kadzo, Ireri and Koech2004) or environment-related factors are more important in determining the well-documented over-dispersion in schistosome populations, but does not in itself discount an importance for parasite genetics. In particular, interactions between host and parasite genetics, or facultative differences due to variations in gene expression, will not be detected by this approach. In addition, high gene flow between parasite populations, as has been observed, will tend to break up familial patterns such that associations with patterns of clinical outcome will be more difficult to detect since frequent recombination through sexual reproduction leads to breakdown of associations between loci. Larger studies, incorporating studies of morbidity as well as intensity, would be needed to equivocally demonstrate a lack of importance of schistosome genetics on clinical outcome, and this study will provide useful parameters for the design of such studies if desired. Interestingly, although Brouwer et al. (Reference Brouwer, Ndhlovu, Wagatsuma, Munatsi and Shiff2003) found genetic differences in RAPD data between parasite populations from children with varying severity of lesions measured by ultrasonography, there were no differences between those with varying infection intensity. An important observation in this study was that the parasite populations of the small number of children who had very high infection intensities, were more likely to contain rare or exclusive alleles. Rare alleles are important for the potential development of drug resistance, and this suggests that targeted treatment of heavily infected individuals might have benefits with regard to managing drug resistance, in addition to the obvious clinical benefits.
In separate analyses we used sibship analysis to estimate the number of familial clusters in the populations of individual children, as well as to estimate the reproductive success of parasite genotypes. We found that this gave us potentially important new insights. Most importantly, we documented for the first time, variable reproductive success of parasite genotypes in an endemic focus. Consistent differences in reproductive success of individual sibships across hosts suggested that at least some of the variation was due to genetic differences, which is a pre-requisite for evolution under selection.
We also demonstrated complex relationships between parasite diversity and infection intensity indicative of phenotypic variation in parasite reproductive success in hosts of different ages and genders. The numbers of adult genotypes were inferred to be higher at Dougouba School, where the average age was higher, than at Bambougou-Wèrè School. This was perhaps unexpected since infection intensity, as measured by egg output is thought to reduce with child age, due the development of acquired immunity. Instead, our results suggest that although egg output may decrease, the diversity of adult worm genotypes present does not. Due to the life cycle of schistosomes, where clonal amplification takes place in the intermediate hosts such that human hosts may contain multiple identical adult worm gentoypes, it could be that the number of adult worm genotypes present is not an accurate reflection of worm burden. Thus their increased diversity perhaps reflects the longer period over which older children could have acquired infections. Indeed, with the exception of the most heavily infected children, the numbers of adult genotypes across ages and schools were higher in the children with lighter intensity infections, such that fitness of individual parasite genotypes (measured as the number of offspring per genotype) was significantly reduced in the more diverse infections of older compared to younger children. This could be explained by there being fewer worms of each genotype present in older children, direct resource competition between these parasite genotypes and/or that acquired immunity may be reducing fecundity.
Similarly, the numbers of adult genotypes were inferred to be higher in boys than girls, suggesting that boys, perhaps through exposure-related behaviour, or innate susceptibility, also have more diverse adult worm infections. Thus our results agree with those of Brouwer et al. (Reference Brouwer, Ndhlovu, Wagatsuma, Munatsi and Shiff2003) who found higher diversity in the S. haematobium populations of boys than girls in Zimbabwe. They also documented that morbidity, as measured by ultrasonography, was more common in boys but that it was not a direct relationship with intensity. We found little evidence that the larval populations of boys were more diverse than those of girls, despite their more diverse adult infections, instead finding higher levels of inbreeding. One potential explanation could be that differences in water contact (sites, activities, duration) may increase the transmission of larger numbers of clonal schistosomes to boys, which would result in increased inbreeding. The reproductive success of parasite genotypes was also reduced in boys, potentially indicative of higher worm burdens and density dependence.
Population genetic studies such as these provide parameter estimates that would be useful for mathematical models aimed at predicting the likely development, spread and control of drug resistance. Our data suggest that non-random mating, and age and gender-based differences should be incorporated into such models. Additionally, the most heavily infected children, which in this study represented about 6% of children, appeared to be unusual and showed no evidence of reduced parasite success (number of offspring per parasite genotype) in more diverse infections. They also contained more rare alleles.
Concluding remarks
This study reports the development of novel tools for the molecular epidemiological analysis of S. haematobium and closely related parasites. We assayed the utility of these tools to describe the population genetic characteristics of an untreated parasite population of 47 children in a geographically compact area of Mali. These data will provide a baseline against which to measure the effect of mass chemotherapy with praziquantel on parasite population genetics in this region and further study this important parasite throughout its distribution.
ACKNOWLEDGEMENTS
We are very grateful to the children and teachers of Bambougou-Wèrè and Dougouba Schools, to the technical team of the Institut National de Recherché en Santé Publique and collaboration with Professor Alan Fenwick and the ongoing SCI programme for enabling sample collections, Dr Alice Norton for molecular assistance and Julia Llewellyn-Hughes for assistance with genotyping reactions.
FINANCIAL SUPPORT
This research was funded by grants from the Wellcome Trust (C.M.G., D.R., J.P.W. Grant no. GR063774), the Royal Society (C.M.G. and J.P.W.), and the Bill and Melinda Gates Schistosome Control Initiative (J.P.W.).