INTRODUCTION
Molecular tools have tremendous potential for providing insights into the biology of parasitic organisms (Criscione et al. Reference Criscione, Poulin and Blouin2005) and have been applied to parasite phylogeography (e.g. Perkins, Reference Perkins2001), parasite dispersal (e.g. McCoy et al. Reference McCoy, Boulinier and Tirard2005), and parasite acquisition (e.g. Walker et al. Reference Walker, Prodohl, Fletcher, Hanna, Kantzoura, Hoey and Trudgett2007). Parasite occurrence patterns and genetic structure can also be used for inferring host population history as parasites generally have shorter generation times, and therefore evolve more rapidly than their hosts (Whiteman and Parker, Reference Whiteman and Parker2005). Given that some parasitic organisms are pathogenic or serve as vectors for pathogenic microorganisms, the use of molecular tools can be especially valuable in tracking pathogen movement and making inferences and predictions about pathogen dynamics.
Yersinia pestis is a zoonotic pathogen whose transmission is primarily dependent on fleas (see Perry and Fetherston (Reference Perry and Fetherston1997) and Gage and Kosoy (Reference Gage and Kosoy2005) for reviews of Y. pestis biology). The aetiological agent for plague, Y. pestis is a globally occurring pathogen that naturally infects over 200 mammalian species, yet its ecology and transmission dynamics are poorly understood (Perry and Fetherston, Reference Perry and Fetherston1997; Gage and Kosoy, Reference Gage and Kosoy2005; Webb et al. Reference Webb, Brooks, Gage and Antolin2006). Effects of Y. pestis infection are highly variable within and among host species; some individuals and some species show high resistance to infection whereas others are highly susceptible (Gage and Kosoy, Reference Gage and Kosoy2005). Plague may be maintained in an enzootic cycle, consisting of small rodents and their fleas, that periodically shifts to epizootic or amplifying, hosts causing massive and rapidly moving die-offs before returning to the enzootic cycle (Gage and Kosoy, Reference Gage and Kosoy2005). Mammalian host species responsible for maintaining Y. pestis are unidentified in most systems, as are the flea species responsible for inter- and intraspecific transmission (Krasnov et al. Reference Krasnov, Shenbrot, Mouillot, Khokhlova and Poulin2006; Eisen et al. Reference Eisen, Bearden, Wilder, Montenieri, Antolin and Gage2006). Prairie dogs (Cynomys) are among the mammalian species most severely affected by Y. pestis often suffering mortality in excess of 99% (Cully and Williams, Reference Cully and Williams2001). Although plague is thought to be primarily a flea-transmitted disease, specific mechanisms to account for plague transmission during epizootic events are unclear (Eisen et al. Reference Eisen, Bearden, Wilder, Montenieri, Antolin and Gage2006; Webb et al. Reference Webb, Brooks, Gage and Antolin2006).
In Boulder County, Colorado, epizootic plague events affecting the majority of black-tailed prairie dog (Cynomys ludovicianus) colonies occur roughly every 5 to 10 years, although the likelihood of plague reaching a particular colony may be influenced by the landscape context in which the colony exists (Collinge et al. Reference Collinge, Johnson, Ray, Matchett, Grensten, Cully, Gage, Kosoy, Loye and Martin2005). Transmission of Y. pestis among hosts is probably dependent on the abundance and prevalence of vector-competent flea species (Lorange et al. Reference Lorange, Race, Sebbane and Hinnebusch2005). Black-tailed prairie dogs in Colorado are primarily parasitized by 2 flea species, Oropsylla hirsuta and O. tuberculata cynomuris (Brinkerhoff et al. Reference Brinkerhoff, Markeson, Knouft, Gage and Montenieri2006), both of which are capable of transmitting Y. pestis (Wilder et al. Reference Wilder, Eisen, Bearden, Montenieri, Tripp, Brinkerhoff, Gage and Antolin2008). Flea parasitism on prairie dogs is seasonal with O. hirsuta reaching peak abundance in summer months and O. t. cynomuris reaching peak abundance in February and March (Wilder et al. Reference Wilder, Eisen, Bearden, Montenieri, Tripp, Brinkerhoff, Gage and Antolin2008). Fleas often have anatomical adaptations suited to their primary hosts (Marshall, Reference Marshall1981; Traub, Reference Traub, Kim and 1985) and, in our study system, tend to exhibit host-specificity (Brinkerhoff, Reference Brinkerhoff2008); O. hirsuta and O. tuberculata are rarely collected from mammalian species other than black-tailed prairie dogs (Brinkerhoff, Reference Brinkerhoff2008; Salkeld and Stapp, Reference Salkeld and Stapp2008).
Substantial reductions in prairie dog population size resulting from plague should negatively affect populations of highly host-specific parasites, such as fleas, which are not prone to utilizing other host species (Brinkerhoff, Reference Brinkerhoff2008; Salkeld and Stapp, Reference Salkeld and Stapp2008). Therefore, the population genetic structure of prairie dog fleas may be reflective of prairie dog population genetic structure. Additionally, flea migration and movement events inferred from molecular data could be used as a proxy for prairie dog migration and movement. A study of seabirds and their ticks revealed insights into host population dynamics and migratory patterns (McCoy et al. Reference McCoy, Boulinier and Tirard2005) and movement of guinea pigs was inferred from a study of the population genetic structure of guinea pig fleas in Peru (de la Cruz and Whiting, Reference de la Cruz and Whiting2003). To elucidate patterns and processes of inter- and intraspecific plague transmission, we explored whether population genetic structure of O. hirsuta (1) is consistent with the hypothesis that fleas are dispersal-limited and (2) shows signals of recent plague events. Specifically, we predicted that genetic differentiation of flea populations would be highest among prairie dog colonies that are farthest apart. We also predicted that colonies with a recent history of plague would show genetic signals of population expansion.
MATERIALS AND METHODS
Study sites and species
We collected fleas from live-trapped black-tailed prairie dogs (Cynomys ludovicianus) at 11 study colonies in Boulder County, Colorado (Fig. 1). C. ludovicianus is a social and colonial burrowing ground squirrel native to west-central North America, east of the Rocky Mountains. Flea assemblages of C. ludovicianus are dominated by Oropsylla hirsuta and O. tuberculata cynomuris (Brinkerhoff et al. Reference Brinkerhoff, Markeson, Knouft, Gage and Montenieri2006), which make up roughly 98% of all prairie dog fleas in this study system (Brinkerhoff, Reference Brinkerhoff2008). All study colonies (Fig. 1) were between 1550 and 1900 meters in elevation and were generally similar in area and vegetation (Conlin, Reference Conlin2005). Plague history was determined in consultation with local land and wildlife managers who have records of plague activity in Boulder County dating from the mid-1980 s (Collinge et al. Reference Collinge, Johnson, Ray, Matchett, Grensten, Cully, Gage, Kosoy, Loye and Martin2005; Markeson, Reference Markeson2005). Colonies included in the plague-positive group are known to have been affected by a major plague epizootic in 1994 whereas plague-negative colonies persisted through this epizootic. Another widespread epizootic affecting most colonies in the study system occurred in 1986 and, though plague history is unknown for some colonies, colonies 18A and 60 A (Fig. 1) are known to have survived.

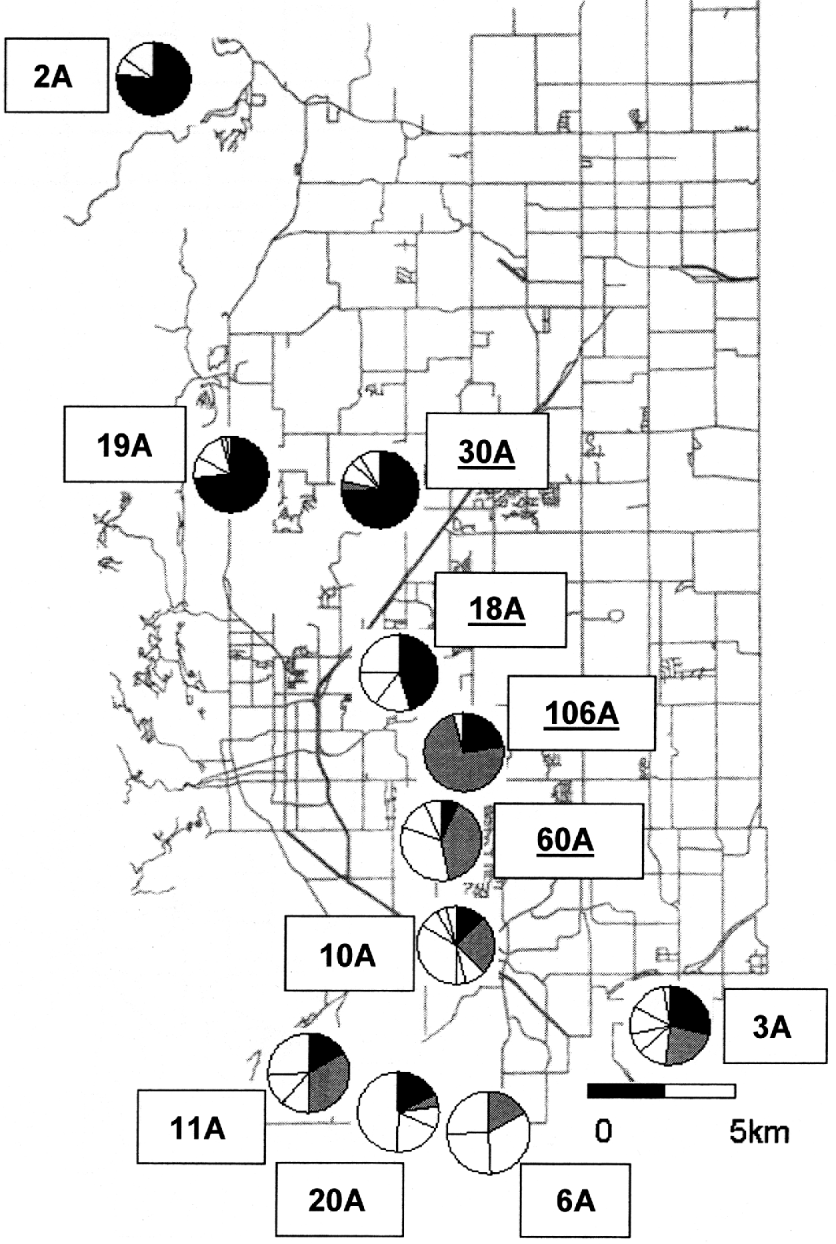
Fig. 1. Locations of sampling sites from which Oropsylla hirsuta samples were collected. Open circles represent sites that escaped a widespread plague epizootic in 1994 and are referred to in the text as ‘plague-negative’. Filled circles represent sites that experienced this plague event and are referred to as ‘plague-positive.’
Sample collection
Prairie dogs were sampled using Tomahawk live-traps (Tomahawk Live Trap, Inc., Tomahawk, WI, USA) and all trapped individuals were sedated with isoflurane to facilitate flea collection. All visible fleas were collected from each prairie dog using forceps and a fine-toothed comb. Fleas were placed in vials containing either 100% ethanol or a 2% saline solution and a small amount of the surfactant Tween (polysorbate 80). The vials were stored at –80°C and the fleas were later identified to species by light microscopy using dichotomous keys presented by Hubbard (Reference Hubbard1968) and Furman and Catts (Reference Furman and Catts1982) or based on COII DNA sequence data.
PCR amplification and sequence analysis
At all colonies except 3A, 6A, 11A, and 19A, we selected 1 flea from each captured host individual for genetic analysis. At colonies 3A and 19A, we selected 1 flea per host from 25 and 31 hosts, respectively. At these sites, we also analysed an additional 11 and 18 fleas, collected from 5 hosts (site 3A) and 4 hosts (site 19A). Sampling at the remaining sites was conducted such that 48 fleas, collected from a total of 10 hosts (range: 1–14 fleas per host) were analysed from site 6A, and 53 fleas, collected from 8 hosts (range: 1–20 fleas per host) were analysed from site 11A (Table 1). DNA from all fleas was extracted using a DNeasy Blood and Tissue Kit (Qiagen Inc., Valencia, CA, USA) following the included standard protocols. Polymerase chain reaction (PCR) was used to amplify a portion of the cytochrome oxidase II (COII) gene using primers Fleu and Rlys (Whiting, Reference Whiting2002). We used Amplitaq Gold LD (Applied Biosystems, Inc., Foster City, CA, USA) DNA polymerase and the following PCR conditions: 95°C for 12 min and 37 cycles of 94°C for 45 sec, 42°C for 45 sec and 68°C for 2 min, followed by a final elongation step of 7 min at 68°C. PCR product was purified with ExoSap (USB Corp., Cleveland, OH, USA) solution and sequencing was performed on an ABI 3730×l with BigDye chemistry 3.1. Sequence data were also collected from 2 individuals of the congeneric flea species O. tuberculata cynomuris for use as an outgroup in phylogenetic analysis.
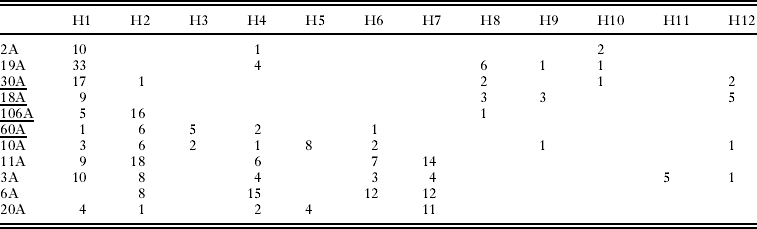
Table 1. Numbers of Oropsylla hirsuta tested, number of prairie dogs sampled, and number of unique COII haplotypes detected at each of 9 study sites in Boulder County, Colorado

Raw sequence data were edited with Sequencher software and were aligned with Clustal X. Unique haplotypes and redundant sequences were identified using MacClade 4.08. To ensure monophyly of O. hirsuta in our sample, we constructed a Neighbor-joining phylogram, rooted with O. t. cynomuris sequence data and following the Kimura two-parameter model of measuring DNA sequence divergence using PAUP 4.0. We used Arlequin 2.0 (Schneider et al. Reference Schneider, Roessli and Excoffier2000) to calculate pairwise Fst values between all pairs of sampling sites and to perform an exact test of population differentiation, done by comparing the observed distribution of haplotypes among populations to those generated from 10 000 Markov steps. Arlequin 2.0 was also used to generate and test mismatch distributions for signals of recent population expansion. We tested the relationship between linearized pairwise Fst and geographical distance using a Mantel test with statistical significance determined by 5000 randomized Monte Carlo runs.
RESULTS
We collected COII sequence data from a total of 332 O. hirsuta (Table 1) sampled from 11 prairie dog colony sites in Boulder County, Colorado (Fig. 1). Sequences collected from all individuals provided clear and unambiguous data consisting of a 480 base-pair section of the COII gene. We identified a total of 24 unique haplotypes, 12 of which were detected at least twice (Table 2). Haplotype H1 was the most common and was detected in 101 individuals sampled at 10 of the 11 sites (Table 2). The second most common haplotype (H2) was detected 64 times and was localized entirely in the southern region of the study area except for 1 detection at site 30A (Table 2, Fig. 2). Several other haplotypes (H3, H5, H6, H7, and H11) were only found in the southern half of the study area whereas 1 haplotype (H10) was only detected in the northern region (Table 2). Two haplotypes only occurred at 1 study area each: H5 was only detected at site 20A and H11 was only detected at site 3A (Table 2).

Fig. 2. Relative abundance of the two most common haplotypes detected (H1 and H2) in this study at each of the 11 study sites. Haplotype H1 is indicated by the black sections of each pie chart and haplotype H2 is indicated by grey sections.
Table 2. Numbers of each Oropsylla hirsuta haplotype detected at 11 prairie dog colonies in Boulder County, Colorado
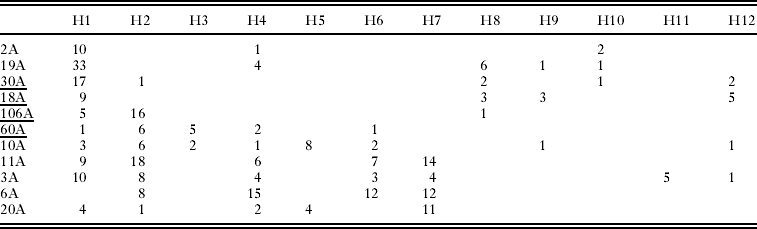
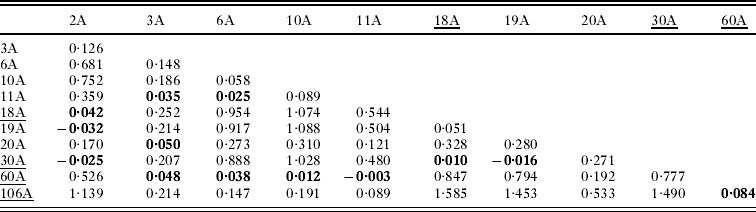
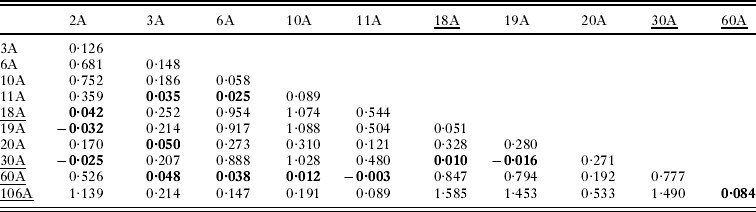
Pairwise linearized Fst values for all pairs of sampling sites are presented in Table 3 and are plotted against the natural log of distance in Fig. 3. Non-differentiation tests show a strong signal of overall population genetic structure (P<0·0001), although non-differentiation could not be rejected (P>0·05) at 13 pairs of sampling sites (Table 3, Fig. 4). The average number of pairwise differences among sequences within populations ranged from 0·92–3·54 (Table 1) and was generally higher in the southern part of the study system. Within-site sequence variation was not dependent on the number of fleas collected from each prairie dog (F=1·67, P=0·23). We detected no statistical evidence of isolation-by-distance in these data (Mantel test P>0·15, r=0·22; Fig. 3). Similarly, there was no evidence of isolation by distance when only pairs of sites that were (P=0·12, r=0·42) and were not (P=0·5, r=0·2) recently affected by plague. Analysis of haplotype mismatch distributions indicated that 3 of the 4 plague-negative sites were different from distributions expected from a recent population expansion model; all of the haplotype distributions from plague-positive sites are consistent with the recent expansion model (Table 1).

Fig. 3. Data representing the relationship between linearized pairwise Fst and geographical distance (Mantel test r=0·2, P>0·1).

Fig. 4. Genetic differentiation among Oropsylla hirsuta populations at black-tailed prairie dog colonies. Lines link populations that were not differentiated by pairwise Fst at alpha 0·05; filled circles indicate historically plague-positive sites and open circles indicate plague-negative sites.
Table 3. Linearized pairwise Fst values of Oropsylla hirsuta collected at 9 prairie dog colonies in Boulder County, Colorado
DISCUSSION
Our results indicate significant population genetic structure in O. hirsuta collected from active prairie dog colonies. The lack of a relationship between pairwise Fst and geographical distance suggests that fleas may not be dispersal limited, at least at the scale of this study. We found evidence to suggest that plague history may influence flea population genetic structure; flea haplotype distributions at all prairie dog colonies that were recently affected by plague were consistent with patterns expected under demographic expansion. Furthermore, the observed pattern of non-differentiation among study sites indicates that clusters of northern and southern colonies are not distinguishable in terms of haplotype distribution, but that there is apparent differentiation between these groups. All of the sites that escaped recent plague events are located in the centre of the study system and may have served as source populations of prairie dogs following die-offs due to plague. Landscape features such as roads and streams may serve as barriers to prairie dogs (Collinge et al. Reference Collinge, Johnson, Ray, Matchett, Grensten, Cully, Gage, Kosoy, Loye and Martin2005; Magle et al. Reference Magle, Theobald and Crooks2009) but are potentially more permeable to other mammalian species that infrequently acquire O. hirsuta, such as desert cottontail rabbits (Sylvilagus audubonii) or coyotes (Canis latrans) (Brinkerhoff, Reference Brinkerhoff2008).
In this study system, periodic plague epizootics result in prairie dog colony extirpations (Collinge et al. Reference Collinge, Johnson, Ray, Matchett, Grensten, Cully, Gage, Kosoy, Loye and Martin2005). All plague-positive colonies in this study are known to have experienced prairie dog die-offs during the same year (1994) and were possibly affected by another widespread epizootic event in 1986 (Collinge et al. Reference Collinge, Johnson, Ray, Matchett, Grensten, Cully, Gage, Kosoy, Loye and Martin2005). Elimination of up to 100% of primary hosts should substantially reduce flea population sizes and the patterns of flea population expansion inferred through mismatch distributions are generally consistent with plague history. All plague-positive colonies show haplotype distributions consistent with recent population expansion, whereas 3 of the 4 plague-negative colonies show haplotype mismatch distributions that differed from the recent expansion model. Among the plague-negative colonies, only 30A and 106A are known to have survived the 1994 plague epizootic whereas colonies 18A and 60A are known to have remained active though the major epizootic events of 1994 and 1986 as well as through smaller-scale epizootics in 1991 and 1999. Thus, the detection of strong departures from the recent expansion model at these sites is consistent with our understanding of spatial plague dynamics in this system.
Geographical distance between prairie dog colonies failed to predict with the degree of genetic differentiation between flea populations, suggesting that distance does not have an important effect on O. hirsuta gene flow in this system. However, we note that rapid expansion of flea populations following plague events would affect migration/drift equilibrium and may reduce the likelihood of detecting isolation by distance. Prairie dog dispersal typically occurs over distances of less than 5 km (Garrett and Franklin, Reference Garrett and Franklin1981) and is negatively affected by urbanization (Magle et al. Reference Magle, Theobald and Crooks2009). Therefore, O. hirsuta may be dispersed by non-prairie dog mammals that move more easily over large distances. Mammalian prairie dog predators are known to acquire prairie dog fleas (McGee et al. Reference McGee, Butler, Pence, Alexander, Nissen, Ballard and Nicholson2006; Salkeld et al. Reference Salkeld, Eisen, Stapp, Wilder, Lowell, Tripp, Albertson and Antolin2007; Brinkerhoff, Reference Brinkerhoff2008), often in the course of predation or foraging activities and O. hirsuta have been collected from desert cottontail rabbits (Sylvilagus audubonii) and small rodents in this system (Brinkerhoff Reference Brinkerhoff2008). Jones and Britten (Reference Jones and Britten2010) found discordance between prairie dog and O. hirsuta population genetic structure and suggested that non-prairie dog mammals might transport O. hirsuta among prairie dog colonies in Montana. The plague history of prairie dog colony sites apparently did not influence the relationship between genetic and geographical distance among flea populations, although linearized Fst values were higher, on average, for pairs of plague-negative sites (0·8, N=6 pairs) than for plague positive sites (0·3, N=21 pairs).
Prairie dog and flea populations that persist during plague events are likely to serve as sources for repopulation of extirpated sites. Potential source colonies are located in the central part of this study system (colonies 18A, 60A and 106A). For example, high relative abundance of haplotype H1 at site 18A was also observed at nearby sites showing signs of recent population expansion (19A, 30A and 2A). Such a pattern could have been caused by founder individuals carrying haplotype H1 that dispersed north to newly vacant sites. A similar mechanism could account for the high relative abundance of haplotype H2 at sites 60A, 106A, 10A and 3A. This haplotype was absent or present in very low relative abundance at northern sites and its distribution could be indicative of southward gene flow from colony 18A.
Sample size is certain to influence which haplotypes are detected at a particular site and it is likely that increased sampling effort would lead to detection of additional haplotypes at each site. The detection of uniquely-occurring haplotypes also points to the probable existence of unsampled pools of fleas in this landscape and suggests that haplotype richness in this system is higher than was observed (Chao, Reference Chao1984). It is also possible that these haplotypes were introduced by individual fleas originating from outside the sampling area. Although the data presented here suggest a radiation of fleas from the plague-negative sites in the central part of the study area, fleas from other sources could re-populate a prairie dog colony following a plague epizootic. The presence of such unidentified sources could account for some of the genetic differentiation detected in the southern portion of the study system.
Plague events are likely to reduce flea populations through the reduction of host populations, although some O. hirsuta may be able to persist in the absence of prairie dog hosts. O. hirsuta have been collected from prairie dog burrows up to 1 year after a plague event (Kartman et al. Reference Kartman, Quan and Lechleinter1962). Although there is some evidence for host-switching by O. hirsuta following prairie dog die-offs, these events are uncommon and suggest that O. hirsuta does not commonly parasitize alternate hosts when prairie dogs are unavailable (McGee et al. Reference McGee, Butler, Pence, Alexander, Nissen, Ballard and Nicholson2006; Salkeld and Stapp, Reference Salkeld and Stapp2008). After first feeding, fleas die quickly from starvation, typically in a matter of days or weeks, depending on temperature and relative humidity (Dryden, Reference Dryden1989; Krasnov et al. Reference Krasnov, Khokhlova, Fielden and Burdelova2001, Reference Krasnov, Khokhlova, Fielden and Burdelova2002). However, records exist of fleas persisting for over 365 days without feeding, under certain conditions (Dryden, Reference Dryden1989) and Y. pestis-infected fleas have been recovered more than 1 year after plague-induced local extinction of prairie dogs (Kartman et al. Reference Kartman, Quan and Lechleinter1962), although such longevity is not common (Krasnov et al. Reference Krasnov, Khokhlova, Fielden and Burdelova2002).
Although the relationship between prairie dog die-offs and flea population genetic patterns is fairly straightforward, mechanisms to account for flea dispersal and migration are less clear. Urbanization affects prairie dog movement (Magle et al. Reference Magle, Theobald and Crooks2009) and landscape features have been shown to influence plague occurrence in prairie dogs in this study system. High cover of roads, reservoirs, and streams surrounding prairie dog colonies is associated with lower probability of plague occurrence (Collinge et al. Reference Collinge, Johnson, Ray, Matchett, Grensten, Cully, Gage, Kosoy, Loye and Martin2005). Such features might serve as movement barriers for terrestrial mammals carrying Y. pestis infection or Y. pestis-infected fleas. The results presented in this study are inconsistent with the hypothesis that O. hirsuta is influenced by landscape boundaries or distance, suggesting that rare host-switching events or wind-borne or mammalian transportation of eggs may result in gene flow among O. hirsuta populations. The results of this study suggest that population genetic structure of the primary prairie dog flea and purported plague vector, O. hirsuta, can provide information regarding past epizootic events, but contribute less information in terms of identifying mechanisms of flea dispersal and plague transmission. Further studies of prairie dog flea genetic structure, as well as explorations of alternate mechanisms for long-distance pathogen dispersal, should do much to elucidate patterns and processes of sylvatic plague transmission.
ACKNOWLEDGEMENTS
This study was funded by the NSF/NIH joint program in Ecology of Infectious Diseases (DEB-0224328) and the National Center for Environmental Research (NCER) STAR program of the US-EPA (R-82909101-0). Additional funding was provided by grants to R.J.B. from the Boulder County Parks and Open Space Department, the Department of Ecology and Evolutionary Biology at the University of Colorado, and the Beverly Sears Fund. During this project, R.J.B. also received fellowship support from the Rozella Smith Fund at the University of Colorado. We would like to thank both Boulder City Open Space and Boulder County Parks and Open Space departments for allowing us to conduct this study on their properties.