INTRODUCTION
Multidrug resistant malaria parasites are a serious concern for malaria control and elimination programmes and, with the increase in treatment failures to artemisinin combination therapies (Dondorp et al. Reference Dondorp, Nosten, Yi, Das, Phyo, Tarning, Lwin, Ariey, Hanpithakpong, Lee, Ringwald, Silamut, Imwong, Chotivanich, Lim, Herdman, An, Yeung, Singhasivanon, Day, Lindegardh, Socheat and White2009), new small molecule antimalarials targeting novel biological targets are urgently required (The malERA Consultative Group on Drugs, 2011; Anthony et al. Reference Anthony, Burrows, Duparc, Moehrle and Wells2012).
The mitochondrial electron-transport chain (ETC) of Plasmodium falciparum is an attractive target for chemotherapy because its functional significance varies from that of the equivalent mammalian system (see Fig. 1) (Rodrigues et al. Reference Rodrigues, Lopes and Moreira2010). For example, as malaria parasites satisfy their energy needs mostly by glycolysis, their mitochondrial ETC does not seem to be essential for ATP production via oxidative phosphorylation in contrast to aerobic eukaryotic cells. Rather, it is likely to be essential for the parasite as it acts as a sink for the electrons produced in other critical processes such as de novo pyrimidine biosynthesis (Mather et al. Reference Mather, Henry and Vaidya2007). Plasmodium genome sequencing has revealed that the genes encoding enzymes involved in the pyrimidine biosynthetic pathway have been conserved whereas those involved in the pyrimidine salvage pathway have not (Painter et al. Reference Painter, Morrisey, Mather and Vaidya2007); therefore, generation of intracellular pyrimidine in malaria parasites relies on mitochondrial processes. As our understanding of the parasites mitochondrial pathway develops and selectivity differences between the parasite and the human form become more apparent efforts have focused on exploiting these unexplored parasite processes and identifying new chemical templates and developing these inhibitors into viable clinical candidates with acceptable efficacy, bioavailability, pharmacokinetic and toxicity profiles (Winter et al. Reference Winter, Kelly, Smilkstein, Dodean, Bagby, Rathbun, Levin, Hinrichs and Riscoe2006). For example, the P. falciparum cytochrome b Q o site has unusual structural features that have not been observed in sequence data from other eukaryotes (such as a four residue deletion in the cd2 helix of Q o), which may help drive drug selectivity (Fisher et al. Reference Fisher, Bray, Ward and Biagini2007). In this review we will discuss recent advances in the development of new lead drug candidates that inhibit specific processes along the parasite ETC.

Fig. 1. Mitochondrial electron transport chain showing DHOD, bc1 and PfNDH2 processes.
DISCUSSION
P. falciparum DHOD inhibitors
Plasmodium asexual parasites require a basal mitochondrial proton gradient for the regeneration of DHODH, which is required for de novo pyrimidine biosynthesis as Plasmodium lacks a pyrimidine scavenger pathway (Gardner et al. Reference Gardner, Hall, Fung, White, Berriman, Hyman, Carlton, Pain, Nelson, Bowman, Paulsen, James, Eisen, Rutherford, Salzberg, Craig, Chan, Nene, Shallom, Suh, Peterson, Angiuoli, Pertea, Allen, Selengut, Haft, Mather, Vaidya, Martin, Fairlamb, Fraunholz, Roos, Ralph, McFadden, Cummings, Subramanian, Mungall, Venter, Carucci, Hoffman, Newbold, Davis, Fraser and Barrell2002). Pyrimidines are fundamental building blocks of all cells. Not only are these metabolites essential in the biosynthesis of DNA and RNA, but they are also required for phospholipid and glycoprotein biosynthesis. In contrast to its human host, the malarial parasite is unable to scavenge pre-formed pyrimidines, and therefore relies on de novo pyrimidine biosynthesis exclusively to maintain its pyrimidine stores. This is confirmed by the lack of pyrimidine scavenging enzymes in the genome sequence (Baldwin et al. Reference Baldwin, Farajallah, Malmquist, Rathod and Phillips2002). The absence of ‘back-up’ machinery to scavenge pyrimidines makes the de novo pyrimidine pathway an attractive target for new therapeutic agents.
Dihydroorotate dehydrogenase (DHOD) catalyses the oxidation of dehydroorotate (DHO) to orotate; a key step in pyrimidine biosynthesis (Liu et al. Reference Liu, Neidhardt, Grossman, Ocain and Clardy2000; McLean et al. Reference McLean, Neidhardt, Grossman and Hedstrom2001). DHOD is a flavin mono-nucleotide (FMN)-dependent mitochondrial enzyme which relies on Coenzyme Q (CoQ) to catalyse the reoxidation of the flavin cofactor (Fig. 2).
Fig. 2. Reactions catalysed by the enzyme DHODase. DHO is catalysed by FMN cofactor which is reoxidized by coenzyme Q.
The efficacy of human DHODH inhibitors for the treatment of rheumatoid arthritis has been well-documented in the literature, demonstrating that DHODH is a druggable target (Chen et al. Reference Chen, Ruben and Dexter1986; Herrmann et al. Reference Herrmann, Schleyerbach and Kirschbaum2004; Boa et al. Reference Boa, Canavan, Hirst, Ramsey, Stead and McConkey2005). Importantly, X-ray structures of human dihydroorotate dehydrogenase (hDHOD) and Plasmodium DHOD have highlighted an extensive variation in amino acid sequence between the human and the malarial enzymes (Baumgartner et al. Reference Baumgartner, Walloschek, Kralik, Gotschlich, Tasler, Mies and Leban2006; Phillips et al. Reference Phillips, Gujjar, Malmquist, White, El Mazouni, Baldwin and Rathod2008; Deng et al. Reference Deng, Gujjar, El Mazouni, Kaminsky, Malmquist, Goldsmith, Rathod and Phillips2009), thereby providing the structural basis for the identification of species-specific inhibitors.
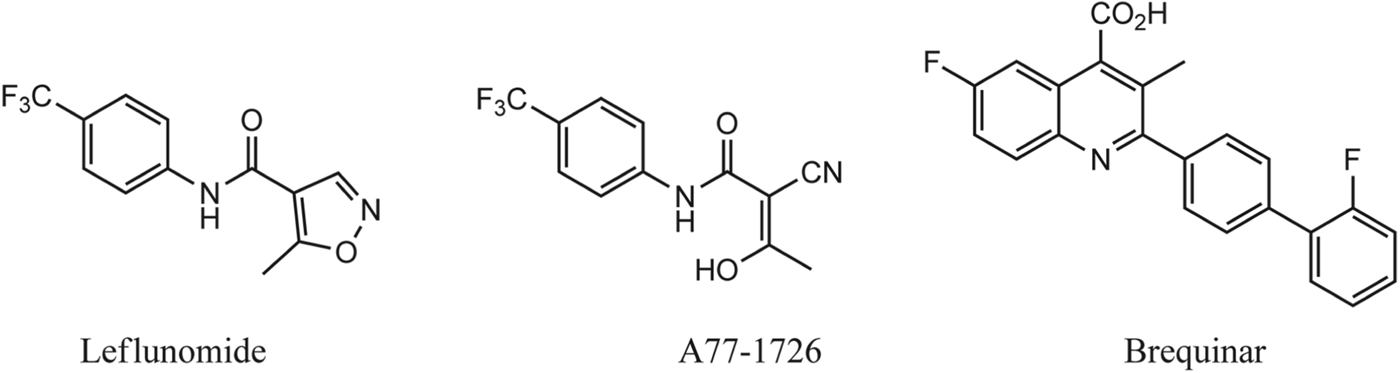
The human version of the enzyme (hDHOD) is the target of a number of inhibitors for the treatment of autoimmune diseases such as rheumatoid arthritis. The potent DHOD inhibitor leflunomide (Arava™, active metabolite A77 1726, Fig. 3) is in clinical use demonstrating that the pathway can be effectively blocked in vivo.
Fig. 3. Potent inhibitors of human DHOD and active metabolite of Lefluomide A77-1726.
Early studies using analogues of potent human DHOD (hDHOD) inhibitors leflunomide and brequinar (Fig. 3) demonstrated poor inhibition of malarial DHOD. The brequinar and A77-1726 binding site observed in the X-ray structure of hDHOD was found to be highly variable between human and malarial enzymes thus confirming that species-selective inhibitor binding was achievable (Malmquist et al. Reference Malmquist, Gujjar, Rathod and Phillips2008).
Although brequinar and A77-1726 have poor inhibition against PfDHOD, the skeletons provided a starting point for drug development of novel PfDHOD inhibitors based on the presumed similarities of the binding pocket. However, early quinoline-based inhibitors designed around the basic brequinar skeleton lacked any inhibitory activity against PfDHOD (Shannon et al. Reference Shannon, Eichholtz, Linstead, Masdin and Skinner1999) and it was evident that a significantly different template would be required for potent species selective inhibition.
The isolation of an X-ray crystal structure of PfDHOD co-crystallized with A77-1726 has allowed for more rational structure-based molecular design of PfDHOD inhibitors (Williamson et al. Reference Williamson, Yea, Robson, Curnock, Gadher, Hambleton, Woodward, Bruneau, Hambleton, Spinella-Jaegle, Morand, Courtin, Sautes, Westwood, Hercend, Kuo and Ruuth1996). A77-1726 was shown to occupy the ubiquinone channel and has a similar positioning of FMN and orotate as shown in the human enzyme. Subtle differences in the dimensions and topography of the hydrophobic ubiquinone channel suggests that PfDHOD is able to accommodate inhibitors that are more cylindrical in overall shape. An approach utilizing the de novo molecular design program SPROUT resulted in molecular templates for the selective inhibition of PfDHOD (Heikkila et al. Reference Heikkila, McConkey, Thirumalairajan, Davies, Parsons, McConkey, Fishwick and Johnson2006).
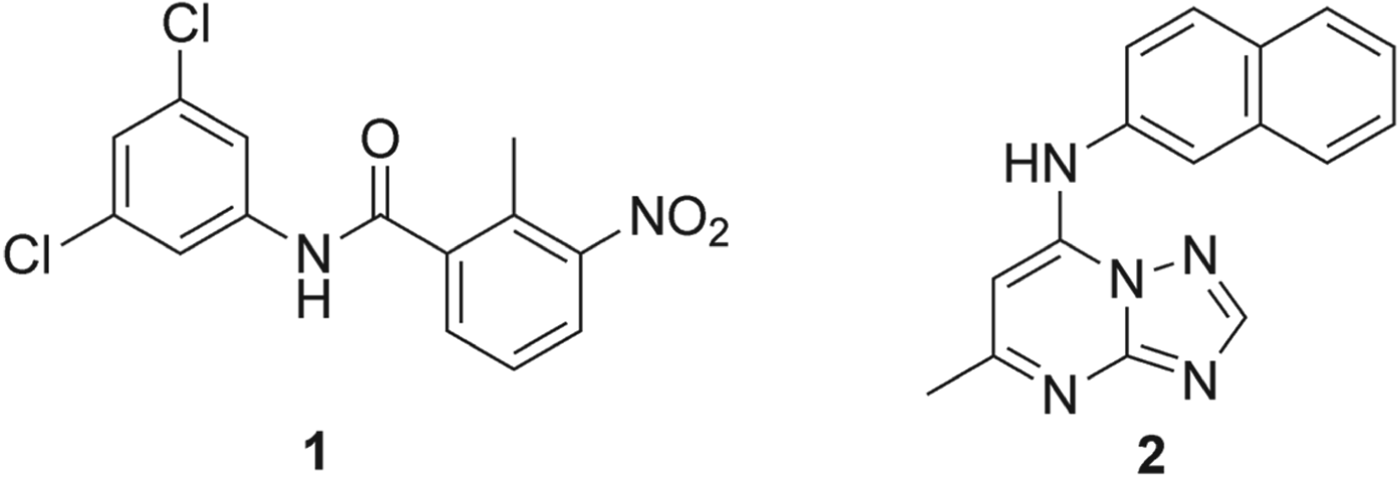
In an attempt to discover novel scaffolds a high throughput screening (HTS) programme was initiated with the aim of screening a diverse library of 220 000 drug-like molecules (Phillips et al. Reference Phillips, Rathod, Baldwin and Gujjar2007). Several classes of PfDHOD inhibitors were identified, including a series of halogenated benzamide/naphthamides and urea-based compounds containing naphthyl or quinolinyl substituents. The most active compound 1 (Fig. 4), displayed potent activity against the isolated enzyme PfDHOD (IC50PfDHOD = 16 nm) and also demonstrated a 12 500 fold difference in specificity against hDHOD.
Fig. 4. New templates for DHOD inhibition.
As noted, the HTS screen by Phillips led to highly selective compounds with potencies below 100 nm against the enzyme (Baldwin et al. Reference Baldwin, Michnoff, Malmquist, White, Roth, Rathod and Phillips2005). Following this initial screen a triazolopyrimidine-based series provided the first example of a DHOD inhibitor with low nanomolar activity against P. falciparum in whole cell-based assays (Patel et al. Reference Patel, Booker, Kramer, Ross, Celatka, Kennedy, Dvorin, Duraisingh, Sliz, Wirth and Clardy2008; Phillips et al. Reference Phillips, Gujjar, Malmquist, White, El Mazouni, Baldwin and Rathod2008; Gujjar et al. Reference Gujjar, Marwaha, El Mazouni, White, White, Creason, Shackleford, Baldwin, Charman, Buckner, Charman, Rathod and Phillips2009). Compound 2, (Fig. 4), was identified as the most promising lead candidate; drug-like, simple and easy to synthesize. However, when administered orally to mice infected with Plasmodium berghei, 2 had no anti-parasite activity. The lack of efficacy of 2 was traced to rapid and possible induced metabolism and to species differences in inhibitor potency between PfDHODH and P. berghei dihydroorotate dehydrogenase (PbDHODH) (Gujjar et al. Reference Gujjar, El Mazouni, White, White, Creason, Shackleford, Deng, Charman, Bathurst, Burrows, Floyd, Matthews, Buckner, Charman, Phillips and Rathod2011).
To provide an insight into the structural basis for potency and species selective binding of malaria-specific inhibitors, the first X-ray crystal structures of PfDHOD bound to triazolopyrimidine inhibitors were reported (Coteron et al. Reference Coteron, Marco, Esquivias, Deng, White, White, Koltun, El Mazouni, Kokkonda, Katneni, Bhamidipati, Shackleford, Angulo-Barturen, Ferrer, Jimenez-Diaz, Gamo, Goldsmith, Charman, Bathurst, Floyd, Matthews, Burrows, Rathod, Charman and Phillips2011). The conformational flexibility of triazolopyrimidines resulted in an unexpected binding mode identifying a new hydrophobic pocket on the enzyme. The malarial enzyme has the flexibility to form two different inhibitor-binding pockets (Pf-naphthyl pocket and the Pf-A77 phenyl pocket). This flexibility may explain why the enzyme can accommodate a number of diverse chemical scaffolds.
Although it was previously thought that inhibitors bind to the ubiquinone (CoQ) site it is now believed that species-selective inhibitors may act by either blocking the electron path between flavin and CoQ or by stabilizing a conformation that excludes CoQ binding. This was demonstrated by introducing a series of mutations in the species-selective inhibitor site. This finding has important implications in future drug design of potent inhibitors since inhibitors can be designed which are chemically different to either substrate (DHO or CoQ) and can be species selective. In addition, because the inhibitor binding site does not overlap with the substrate, it is less constrained to be conserved in amino acid sequence (Gujjar et al. Reference Gujjar, El Mazouni, White, White, Creason, Shackleford, Deng, Charman, Bathurst, Burrows, Floyd, Matthews, Buckner, Charman, Phillips and Rathod2011).
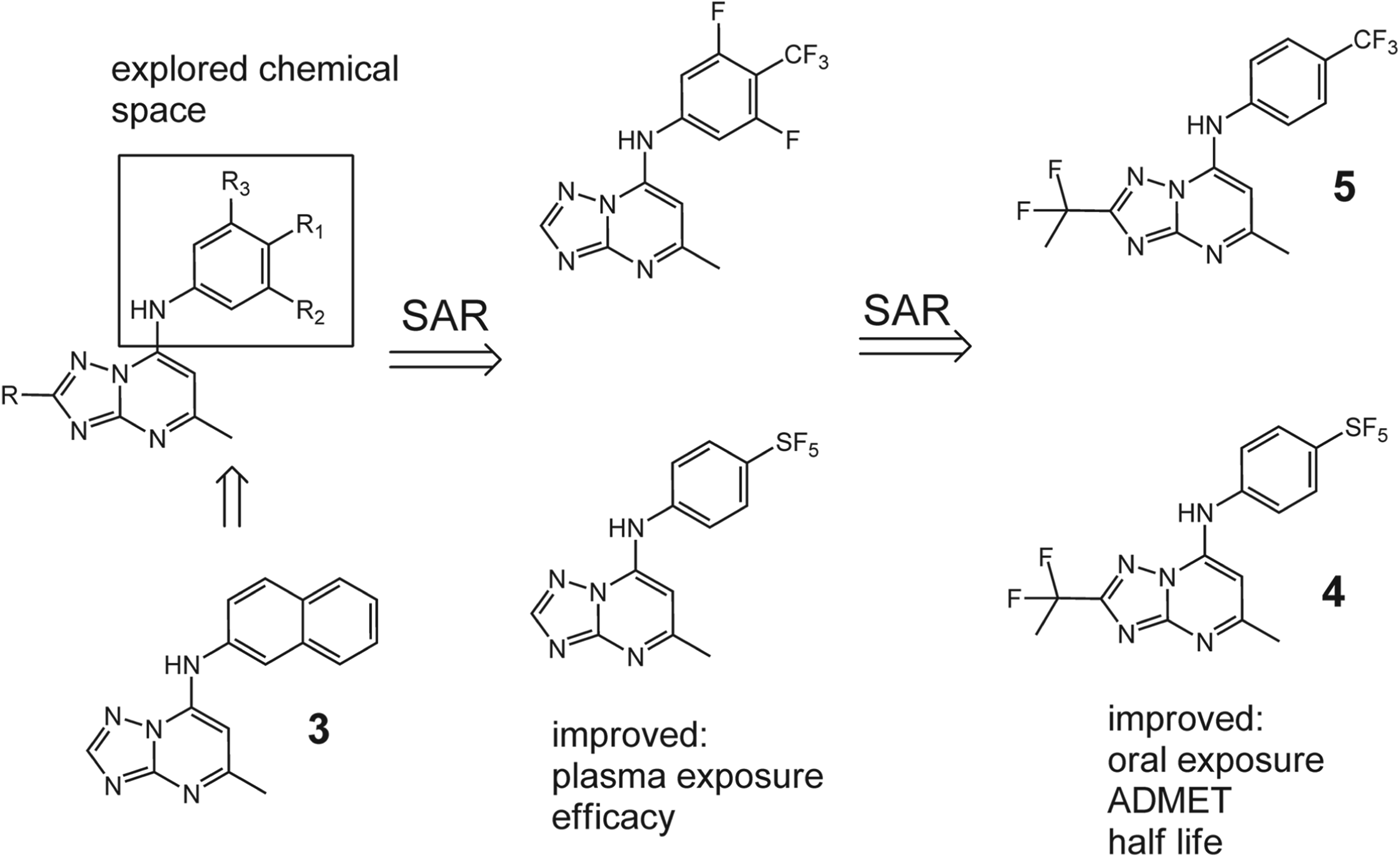
Exploration of a variety of aryl, alkyl and alicyclic/aralkyl substituents linked to the triazolopyrimidine scaffold through the pendant nitrogen (N1) (Fig. 5) exhibited a close correlation between the potency of compounds as inhibitors of PfDHODH and their activity against P. falciparum 3D7 cells. The most potent analogues in the series were naphthyl (structure 3) and 4-tert-butyl-phenyl analogues. The best of these compounds had IC50 values against PfDHODH in the 30 nm range. The next most potent analogues expressed mid-range IC50 values (200–400 nm) and contain a hydrocarbon of less than five carbons or a CF3 or SF5 group at the para position of the aniline ring, with addition of a meta-fluorine providing an incremental increase in compound potency. Larger para substituents led to reduced activity and most of the other characterized compounds were substantially less potent. This observed structure-activity relationship (SAR) is consistent with structural studies, which demonstrate the binding pocket to be entirely hydrophobic. Substitution of 4-SF5 for 4-CF3 resulted in a compound 4 with 2–3-fold better activity against PfDHODH than 2, possibly due to the increase in hydrophobicity with SF5 as compared to CF3. The addition of a meta-fluorine to the aniline ring was tested in combination with some of the better para substituents (CF3 or tert-butyl) and provided somewhat improved potency in each case (Fig. 5).
Fig. 5. Development of late lead triazolopyrimidines 4 and 5 through multiphase SAR studies.
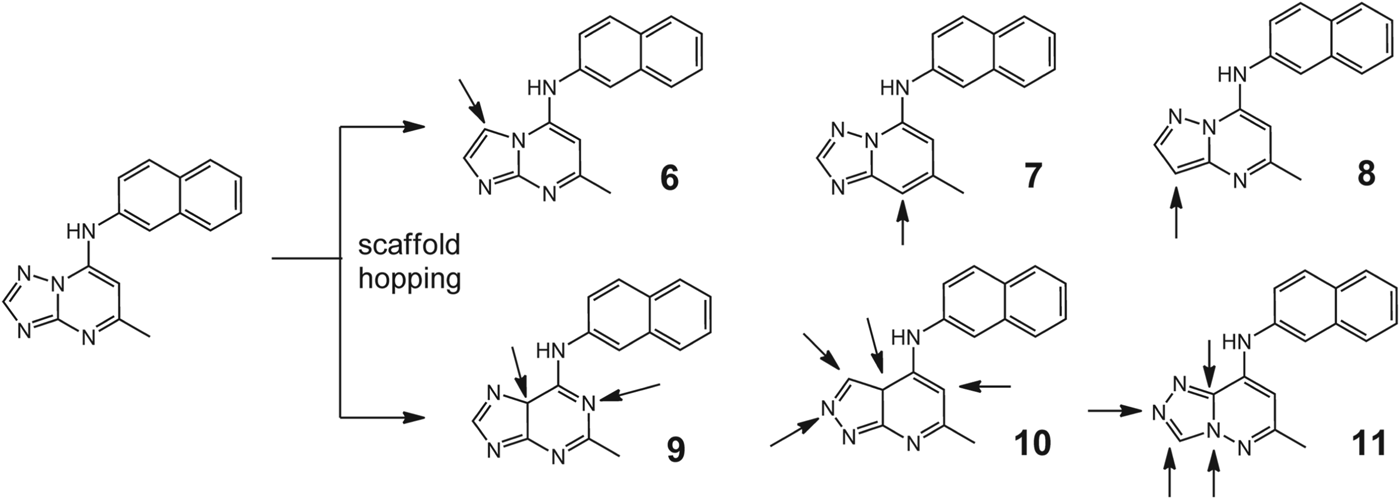
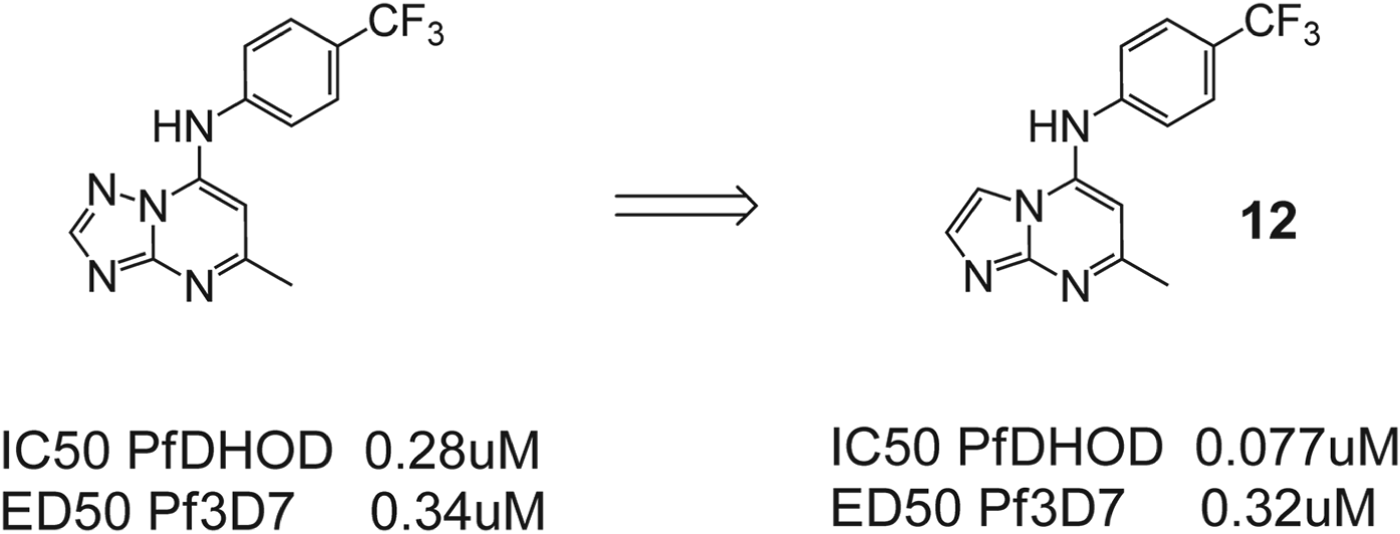
All compounds tested in the series that showed activity against PfDHODH retained good selectivity over human dihydroorotate dehydrogenase (hDHODH). All of the compounds examined had molecular weights of <400, a polar surface area of <85 Å2, number of hydrogen bond donors of <2 and acceptors (e7), and number of freely rotating bonds (e5), consistent with the ranges typically targeted for good oral absorption. Plasma protein binding was in the range of 87–95% for the majority of compounds. As part of a back-up programme to identify additional compounds with clinical candidate potential, the authors extended their exploration of the triazolopyrimidine scaffold by applying bioisosteric morphing, a hit optimization strategy to replace the core of a bioactive compound with groups of similar physical or chemical properties without loss of activity (Marwaha et al. Reference Marwaha, White, El_Mazouni, Creason, Kokkonda, Buckner, Charman, Phillips and Rathod2012). Using this strategy, they synthesized a series of compounds with heteroatom rearrangements/replacements in the triazolopyrimidine ring (6–11, Fig. 6). Each new ring system initially possessed the naphthyl amine (as in 1), but then, for active scaffolds, additional aryl amines were synthesized to determine if the optimal group at that position remained similar to what was observed for the triazolopyrimidines. Three additional heteroatom ring configurations were identified that showed sub-micromolar PfDHODH activity (6, 7 and 9), with the best being the imidazo[1,2-a]pyrimidines (6 and 12, Fig. 7) having equivalent to more potent binding affinity as compared to the matched triazolopyrimidine. The imidazo[1,2-a]-pyrimidine 12 was chosen for profiling of its in vivo properties, including pharmacokinetic analysis and efficacy testing in the P. berghei mouse model. Plasma exposure was lower than for the matched triazolopyrimidine, leading to reduced efficacy. The in vitro metabolism data suggested that the imidazo[1,2-a]-pyrimidine scaffold is more extensively metabolized.
Fig. 6. Biosteric transformations applied to identify pharmacophore template requirements for activity.
Fig. 7. Imidazopyrimidine scaffold with enhanced PfDHOD activity.
Compound 7, in which the pyridine nitrogen N5 is replaced with carbon, was 100-fold less potent than the corresponding triazolopyrimidine (1), demonstrating that the interaction between the pyrimidine nitrogen N5 and the binding site is essential for high-affinity binding (Patel et al. Reference Patel, Booker, Kramer, Ross, Celatka, Kennedy, Dvorin, Duraisingh, Sliz, Wirth and Clardy2008; Gujjar et al. Reference Gujjar, El Mazouni, White, White, Creason, Shackleford, Deng, Charman, Bathurst, Burrows, Floyd, Matthews, Buckner, Charman, Phillips and Rathod2011).
These findings have encouraged the de novo molecular design of various inhibitors. In parallel to this, one such programme has focused on generating analogues of the active metabolite (A77 1726) of potent DHODase inhibitor leflunomide (Heikkila et al. Reference Heikkila, McConkey, Thirumalairajan, Davies, Parsons, McConkey, Fishwick and Johnson2006). The overall aim of the study was to improve selectivity of this inhibitor to the cavities of PfDHOD and hDHOD by utilizing the hydrogen bond potential of the ‘head-group’ and modifying the ‘tail’ to fill the hydrophobic pocket at the entrance to the binding pocket (Fig. 8) (Davies et al. Reference Davies, Heikkila, Mc Conkey, Fishwick, Parsons and Johnson2009).
Fig. 8. Inhibitors (13 and 14a, b) generated by SPROUT-LeadOpt drug discovery exhibited enhanced affinity for PfDHOD vs lead compound A77-1726 (Heikkila et al. Reference Heikkila, McConkey, Thirumalairajan, Davies, Parsons, McConkey, Fishwick and Johnson2006) and Genzyme led preclinical candidates 15a and b.
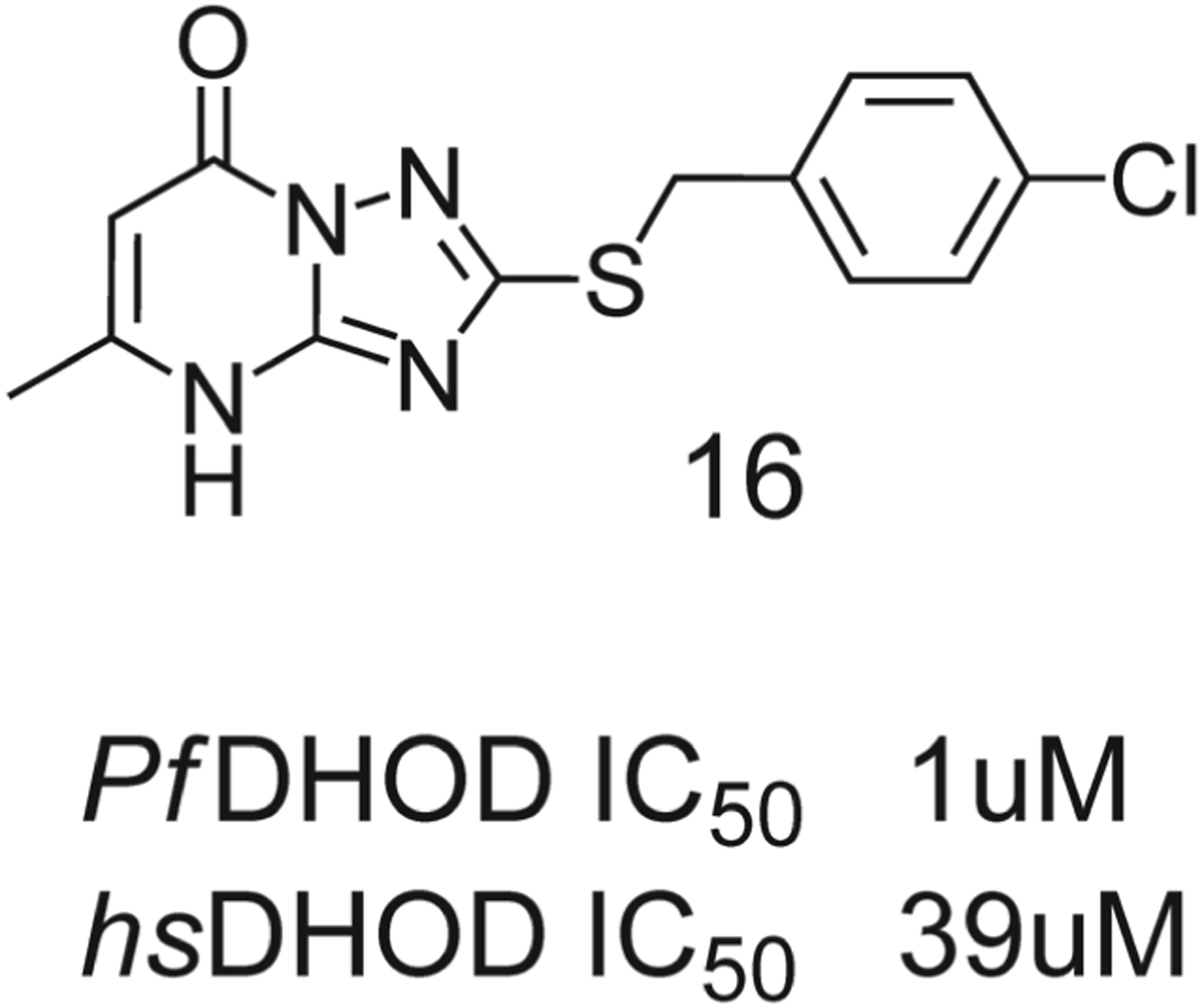
The chemistry was guided using a software package for structure-based drug discovery and lead optimization (SPROUT-LeadOpt) which takes the structure of a small molecule complexed to a protein and generates variants predicted to have higher binding capabilities. Many of the inhibitors synthesized showed enhanced potency (derived experimentally) vs A77-1726. Generally for enhanced potency within this series, a non-planar, substituted biphenyl ‘tail’ was required. Within this region, the shape differences between the hydrophobic region in both human and Plasmodium DHODH can be exploited. A planar nitrile-enamide head group was found to be particularly important for hydrogen bonding; substitution of this hydrogen for methyl removes this intramolecular H-bond and distorts the planarity, disrupting the ligand-enzyme H-bonds. Preliminary testing against parasites identified three compounds, 13 and 14a and b, which exhibited activity in the micromolar range. More recently, Heikkila et al. have used ROCS (rapid overlay of chemical structures) methodology to sample a commercial compound library and develop a series of S-benzyltriazolepyrimidine analogues such as compound 16, Fig. 9, with inhibitor activity, to probe the structural criteria of this class of compounds for selectivity between parasite DHOD and the human form (Heikkila et al. Reference Heikkila, McConkey, Thirumalairajan, Davies, Parsons, McConkey, Fishwick and Johnson2006).
Fig. 9. S-benzyltrazolpyrimidine 16.
In another recent development, a group lead by Genzyme Corp. applied an iterative program, derived from a high-throughput screen of 215 000 compounds from an in-house library, to develop two new PfDHOD inhibitors (15a and b, Fig. 8) with excellent in vivo activity (Booker et al. Reference Booker, Bastos, Kramer, Barker, Skerlj, Sidhu, Deng, Celatka, Cortese, Guerrero Bravo, Crespo Llado, Serrano, Angulo-Barturen, Jimenez-Diaz, Viera, Garuti, Wittlin, Papastogiannidis, Lin, Janse, Khan, Duraisingh, Coleman, Goldsmith, Phillips, Munoz, Wirth, Klinger, Wiegand and Sybertz2010). These compounds, based upon a benzimidazole template, have good oral exposure, are well tolerated at doses up to 200 mg kg−1 day−1and showed in vivo efficacy in P. berghei models, with 4-day ED90 values between 40–56 mg kg−1 day−1. These promising compounds are currently being evaluated for preclinical toxicity to ascertain if they may be suitable as clinical lead candidiates.
In summary, PfDHOD is an attractive target for the design of novel inhibitors. Due to the large differences in amino acids between the binding domains of PfDHOD and hDHOD inhibitor selectivity has been achieved for a diverse range of scaffolds. Although selectivity is desirable, it may not be essential as the human salvage route would replace de novo pyrimidine biosynthesis. For this reason non-species selective DHOD inhibitors continue to be investigated. Knowledge of the proposed inhibitor binding pocket in addition to key interactions validated by site-directed mutagenesis has provided significant SAR to guide future drug design (Bedingfield et al. Reference Bedingfield, Cowan, Acklam, Cunningham, Parsons, Mc Conkey, Fishwick and Johnson2012). The challenge now is to identify inhibitors with a balance of good plasma exposure, oral bioavailability and metabolic stability without decreasing potency vs PfDHOD.
Plasmodium cytochrome bc1 inhibitors
New drugs that target the Plasmodium cyt bc 1 complex, in the parasite ETC, but can circumvent atovaquone resistance or are more robust to resistance would be of great benefit. Currently, several different chemotypes targeting the cyt bc 1 complex have been developed, these include the hydroxynaphthoquinones (atovaquone analogues), pyridones (clopidol analogues), and acridine-related compounds (Valiéres et al. Reference Vallieres, Fisher, Antoine, Al-Helal, Stocks, Berry, Lawrenson, Ward, O'Neill, Biagini and Meunier2012). The cytochrome bc 1 complex is a key mitochondrial enzyme which catalyses transfer of electrons maintaining the membrane potential of mitochondria (Kessl et al. Reference Kessl, Lange, Merbitz-Zahradnik, Zwicker, Hill, Meunier, Palsdottir, Hunte, Meshnick and Trumpower2003). This electron transfer is coupled to the vectorial translocation of protons across the inner mitochondrial membrane.
The proton motive catalytic mechanism of the cyt bc 1 complex is described in detail elsewhere but briefly, the cycle requires two distinct quinone-binding sites (the quinol oxidation site, and the quinone reduction site), located on opposite sides of the membrane and linked by a trans-membrane electron-transfer pathway. Cytochrome b provides both sites and the trans-membrane electron pathway. Quinol antagonists are potent inhibitors of the cyt bc 1 complex, abolishing the proton motive activity of the enzyme and blocking the transfer of electrons to downstream acceptors which collapses the mitochondrial membrane potential and is lethal to P. falciparum. Inhibitors specific to the Q i or Q o sites (such as antimycin (Q i), myxothiazol (Q o) and stigmatellin (Q o)) have been known for many years, and their modes of action are well described (Berry and Huang, Reference Berry and Huang2011). However, these compounds are not species selective (at least not in humans) and are therefore unsuitable for therapeutic use.
Atovaquone is the only established drug in clinical use targeting the P. falciparum cyt bc 1 complex (Nixon et al. Reference Nixon, Moss, Shone, Lalloo, Fisher, O'Neill, Ward and Biagini2013). The rapid emergence of resistance to atovaquone observed during its clinical development resulted in its use in combination with proguanil (Malarone™) (Canfield et al. Reference Canfield, Pudney and Gutteridge1995; Fisher et al. Reference Fisher, Majid, Antoine, Al-Helal, Warman, Johnson, Lawrenson, Ranson, O'Neill, Ward and Biagini2012). The high cost of atovaquone limits its widespread use in resource-poor disease-endemic areas and cheaper alternatives that can overcome resistance are currently being developed.
Here we describe recent advances of cyt bc 1-targeted inhibitors that include pyridones (clodipol analogues), acridone analogues and quinolones (Kelly et al. Reference Kelly, Smilkstein, Brun, Wittlin, Cooper, Lane, Janowsky, Johnson, Dodean, Winter, Hinrichs and Riscoe2009; Barton et al. Reference Barton, Fisher, Biagini, Ward and O'Neill2010; Cross et al. Reference Cross, Monastyrskyi, Mutka, Burrows, Kyle and Manetsch2010, Reference Cross, Maignan, Mutka, Luong, Sargent, Kyle and Manetsch2011; Winter et al. 2006). Significantly, many of these developmental compounds demonstrate little cross-resistance with atovaquone-resistant parasite strains and selected classes have excellent oral activity profiles in rodent models of malaria.
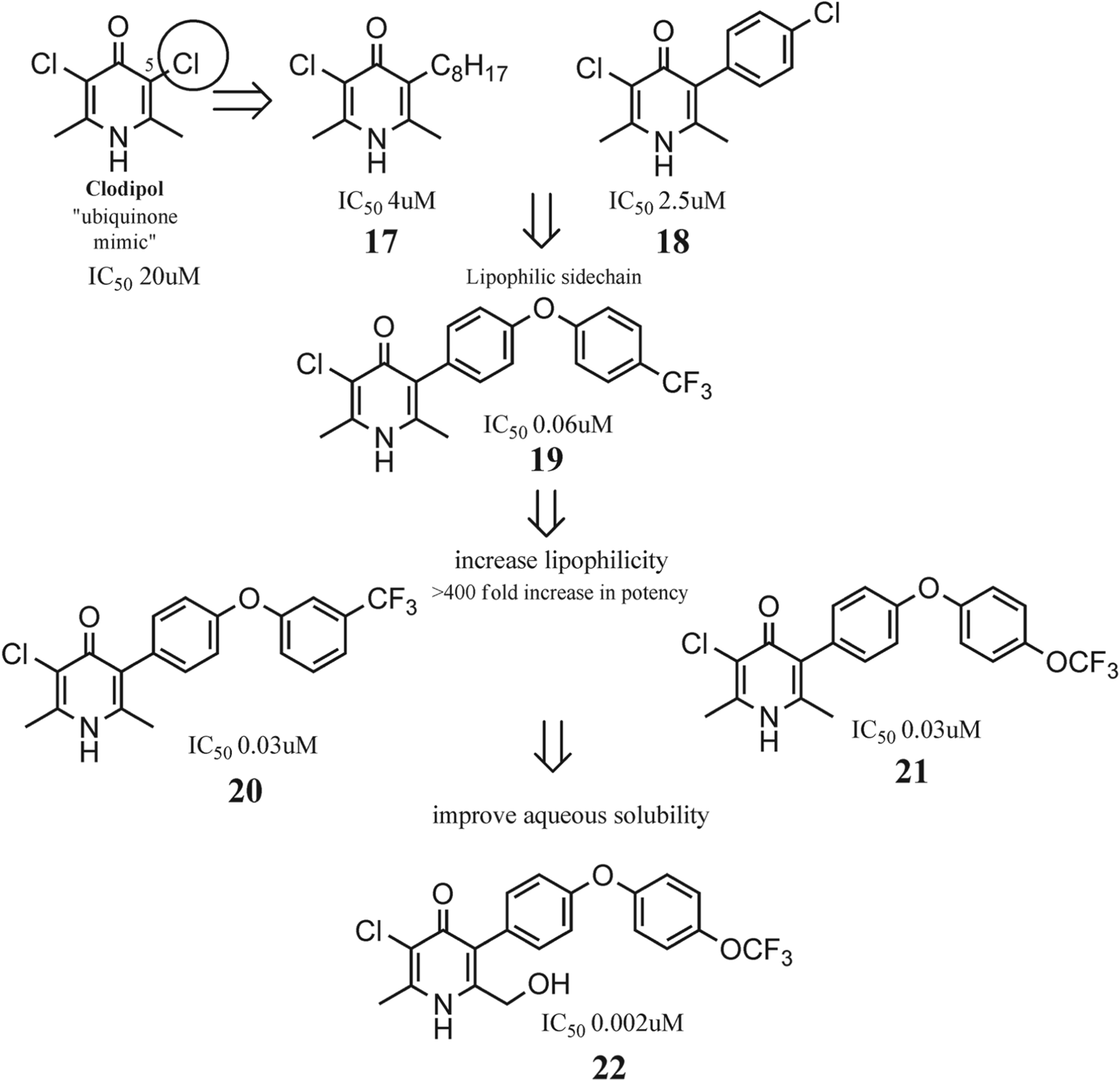
Inhibitors of the plasmodial cytochrome bc 1 such as decoquinate (Nam et al. Reference Nam, McNamara, Bopp, Dharia, Meister, Bonamy, Plouffe Kato, McCormack, Bursulaya, Ke, Vaidya, Schultz and Winzeler2011) have been reported in the literature, demonstrating the potential of mitochondrial inhibition in the development of novel potent therapeutic agents with efficacy not only against blood stages, but also against liver forms and gametocytes. As inhibition of the mammalian respiratory chain could result in serious toxicity, high selectivity against mammalian mitochondria is of paramount importance in ensuring the safety of the compound. Selectivity should be confirmed in vitro both at the level of the target and at the level of the whole cell to ensure that a wide therapeutic index is present in vivo, GSK have reported extensive studies in the development of novel cyt bc 1 inhibitors based upon a pyridone structural template related to the anti-coccidial drug clopidol (see Fig. 10). The original strategy, based on the theory that the 4(1H)-pyridone core was acting as a structural mimic for ubiquinone, focused initially on position C5 by replacing one of the chlorine atoms present at clopidol with different lipophilic moieties (Fig. 10) (Bueno et al. Reference Bueno, Herreros, Angulo- Barturen, Ferrer, Fiandor, Gamo, Gargallo-Viola and Derimanov2012). The introduction of a flexible saturated N-octyl chain, resembling the known antimalarial endochin (Steck, Reference Steck1972; Wiselogle, Reference Wiselogle and Edwards1946) or a 4-chlorophenyl group resulted in 17 and 18, respectively, with modest in vitro antimalarial activity.
Fig. 10. Development of potent plasmodium bc1 inhibitors from a Clodipol template.
It is noteworthy that the introduction of side chains bearing two aromatic rings produced a significant increase in the in vitro activity and more lipophilic groups such as trifluoromethyl instead of a chlorine atom produced 19, with antimalarial activity in vitro in the low nanomolar range against the P. falciparum sensitive 3D7A strain. Significantly antimalarial activity was diminished when the substituent flexibility around the pyridone ring was increased by introducing a methylene group as a linker between the 4(1H)-pyridone ring and the proximal aromatic ring in the lipophilic side-chain, in comparison with their more rigid chlorinated analogues.
The optimization of the 5-phenyloxy-phenyl series produced lead compounds 20 and 21. Derivatives 20 and 21 displayed many sought-after properties for an antimalarial drug: high efficacy in vitro and in vivo in the Plasmodium yoelii murine model and activity against resistant strains to antimalarials including the atovaquone/chloroquine-resistant clones. In addition, 20 and 21 inhibited selectively the plasmodial ubiquinol:cytochrome c oxidoreductase (cyt bc 1) with potencies in the low nanomolar range, similar to the whole-cell in vitro activities seen against P. falciparum cultures. The IC50 for human cyt bc 1 was approximately 150–250-fold higher than the IC50 for Plasmodium cyt bc 1, while the whole-cell selectivity index is >500–1000-fold (measured as cytotoxic IC50 in HepG2 cells vs IC50 for the Pf3D7 strain).
The potent activity observed against atovaquone-resistant strain FCR3A sggests that, despite the similar mechanism of action, the binding modes of the antimalarial 4(1H)-pyridones and atovaquone to the active site of the plasmodial cytochrome bc 1 might not be the same.
On the basis of their good in vivo efficacy in the P. yoelii murine model, compounds 20 and 21 were tested in Aotus monkeys infected with P. falciparum, a primate model similar to human forms. Both 20 and 21 delivered 100% cure after oral administration of seven daily doses of 10 mg kg−1 (Pudney et al. Reference Pudney, Yeates, Pearce, Jones and Fry1992). The oral bioavailability in mice was excellent from solution formulations at low dose (Xiang et al. Reference Xiang, McSurdy-Freed, Moorthy, Hugger, Bambal, Han, Ferrer, Gargallo and Davis2006) (F = 50–100% at <1 mg kg−1), but dropped significantly when administered in suspension at higher doses.
In preclinical safety studies, 21 displayed non-linear pharmacokinetics, limiting exposure levels. No visible signs of toxicity in the repeat dose studies in both rat and dog were observed at the highest exposures. However, the progression of 21 was discontinued because of mild and reversible histopathological findings seen in skeletal and cardiac muscles probably as a result of its high lipophilicity (Dressman and Reppas, 2000) and long half-life in the dog (t 1/2 = 143 h).
A back-up programme was centred on designing novel less lipophilic 5-(4-phenoxyphenyl)-4(1H)-pyridone derivatives, with improved physicochemical properties and oral bioavailability, as well as a shorter half-life in order to prevent possible adverse effects due to accumulation. The programme was focused on reducing lipophilicity by attaching polar hydrophilic groups such as CH2OH or ionizable moieties, such as tertiary amines and carboxyl groups, at the yet unexplored C2 and C6 positions of the 4(1H)-pyridone ring. The introduction of amines, acids or amides at positions C2 or C6 produced similar results to those observed for position C3, leading to inactive compounds (IC50 > 0·25 μ m) irrespective of their relative position in the 4(1H)-pyridone ring (Fig. 10, structure 19a) (Bueno et al. Reference Bueno, Manzano, Garcia, Chicharro, Puente, Lorenzo, Garcia, Ferrer, Gomez, Fraile, Lavandera, Fiandor, Vidal, Herreros and Gargallo-Viola2011). The polar hydroxymethyl group showed a very different behaviour. Thus, while the CH2OH group induced a significant reduction in the in vitro antimalarial activity when located at position C2, with the CH2OH group attached at position C6 the high level of antimalarial activity was retained. 22 displays better physicochemical properties than its precursor 21. The lipophilicity of 22 expressed in terms of logD (Valkó et al. Reference Valkó, Du, Bevan, Reynolds and Abraham2001) is slightly lower in comparison to other pyridones while retaining similar levels of antimalarial activity in vitro and in vivo. In addition, 22 is significantly more soluble in FeSSIF (Fed state simulated intestinal fluid) and less protein bound than 21, with high permeability. In common with 21, hydroxymethyl derivative 22 inhibits plasmodial cytochrome bc 1 in the low nanomolar range, similar to the whole cell in vitro activities seen against P. falciparum cultures. The ratio of mammalian to parasite IC50 for Cyt bc 1 in both human and rat cells (57- and 86-fold, respectively) is slightly lower than for 21 (150-fold).
The higher solubility of 22 resulted in a significant enhancement in oral bioavailability while being more prone to metabolic degradation, possibly by direct elimination via Phase II metabolism as evidenced by the presence of the corresponding glucuronic acid conjugate. In addition, in spite of its shorter half-life, compound 22 displayed an outstanding in vivo efficacy (ED90 = 0·6 mg kg−1) in both P. yoelii and P. falciparum murine malaria models (Jiménez-Díaz et al. Reference Jimenez-Diaz, Mulet, Viera, Gomez, Garuti, Ibanez, Alvarez-Doval, Shultz, Martinez, Gargallo-Viola and Angulo-Barturen2009). However, 22 also demonstrated non-linear pharmacokinetics in preclinical species with decreased oral bioavailability at high doses of the solid dosage form albeit with significantly higher systemic exposures compared to 15 without similar toxic effects.
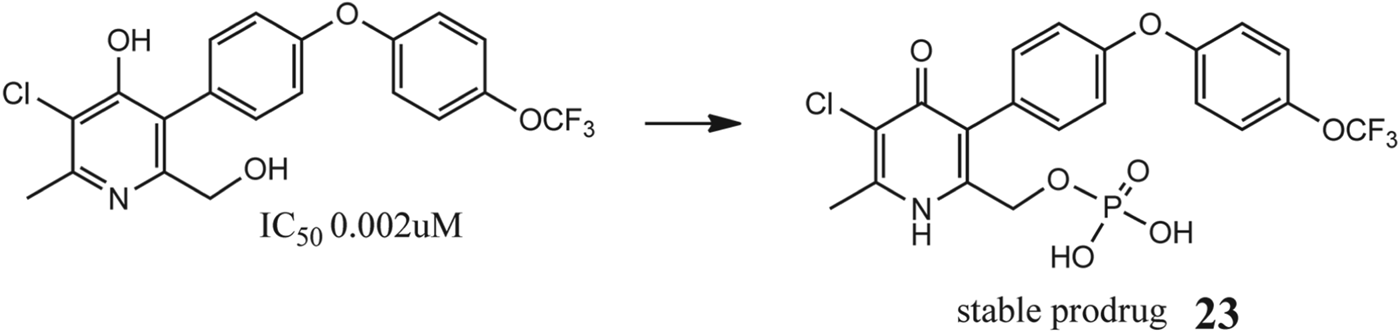
As preclinical studies showed no dose-dependent toxicity, lead candidate 22 was progressed to a first time in human study (FTIH). The requirement for complex formulations to improve oral bioavalability and the resultant significant increase in the associated cost of goods altered the focus towards developing prodrugs of the lead that possessed improved aqueous solubility.
For functionalization purposes 22 has two hydroxyl groups where a suitable pro-moiety could be attached: the C4-OH group (at the hydroxypyridine tautomeric form) and the new primary hydroxyl group C6-CH2OH (Fig. 11). Initial efforts at the C4-OH proved to be unsuccessful, leading to materials that were chemically unstable; however, the phosphate ester prodrug 23 (Fig. 11) was easily accessible and chemically very stable in different aqueous solutions in a wide range of pH and temperatures, remaining unaltered after 48 h (Bueno et al. Reference Bueno, Herreros, Angulo- Barturen, Ferrer, Fiandor, Gamo, Gargallo-Viola and Derimanov2012). Phosphate esters increase solubility, often by several orders of magnitude, by virtue of the di-anionic phosphate group (Furfine et al. Reference Furfine, Baker and Hale2004), and on oral administration, undergo rapid bioconversion at the intestine interface by action of membrane-bound alkaline phosphatases (McComb et al. Reference McComb, Bowers and Posen1979; Heimbach et al. Reference Heimbach, Fleisher, Kaddoumi, Stella, Borchardt, Hageman, Oliyai, Maag and Tilley2007), releasing high concentrations of the parent drug in the vicinity of the mucosal membrane. For optimal absorption, it is required that the poorly soluble parent drug possesses high permeability across membranes as is the case with 22.
Fig. 11. Development of a stable phosphate prodrug from functionalization of the C6-hydroxyl moiety.
PK studies of both 22 and its prodrug 23 were undertaken to examine the effect of increased solubility. The blood levels of parent 22 were significantly higher after administration of its soluble phosphate prodrug 23. As an example, the systemic exposures to 23 were approximately tenfold higher than the maximum exposures reached after administration of 22 in suspension at 1000 mg kg−1. 23 was progressed into preclinical studies as a potential back-up for 22 on the basis of its outstanding physicochemical and pharmacokinetic profile. However, unexpected acute toxicity was observed in rats after oral administration of prodrug 23, which was subsequently replicated by dosing 22 alone and has been attributed to the inhibition of the mammalian cyt bc 1 mitochondrial complex by the parent compound.
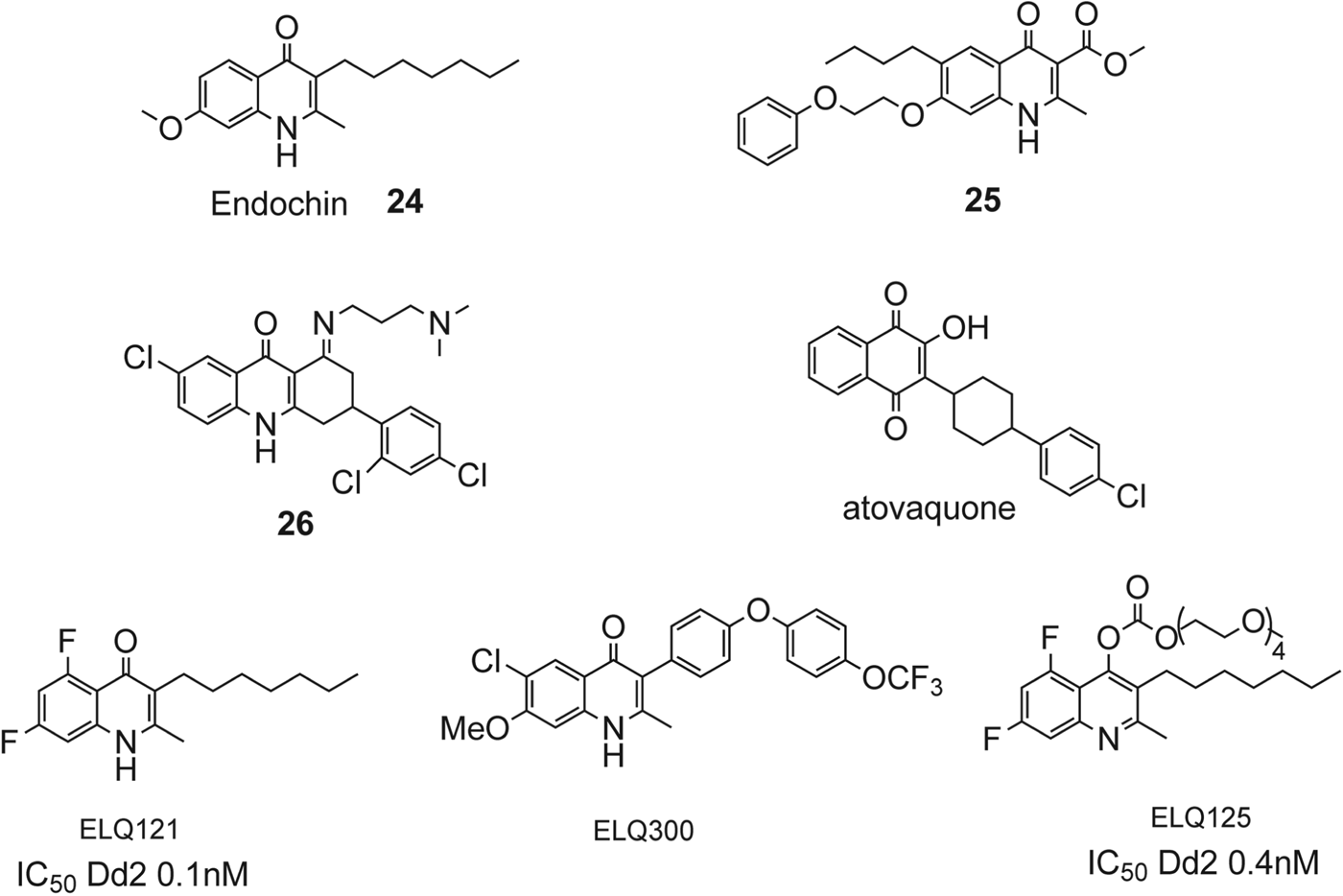
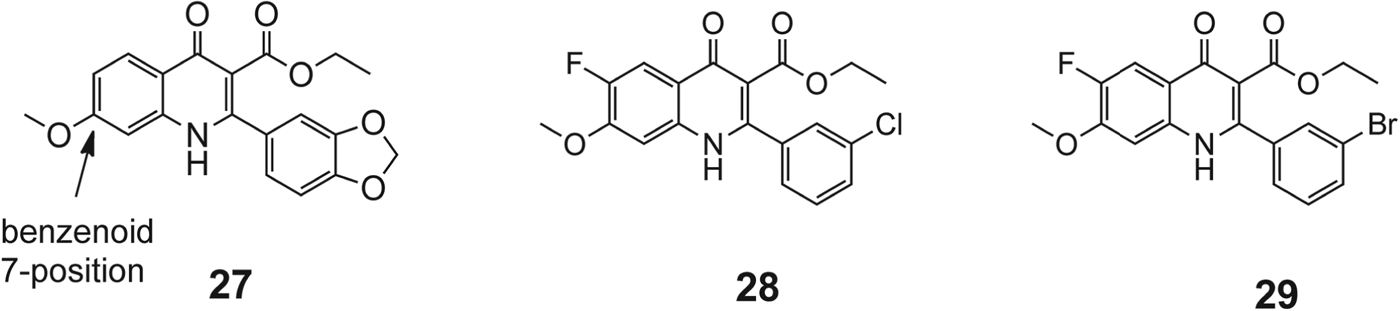
Endochin (Fig. 12, 24) possesses prophylactic and therapeutic activity in avian Plasmodia (Kikuth et al. Reference Kikuth and Mudrowreichenow1947; Salzer et al. Reference Salzer, Timmler and Andersag1948), but lacks activity in mammalian malarial models. The related 4(1H)-quinolone ester 25, discovered in the 1960s, exhibits blood schizonticidal activity against P. berghei in rodents as well as prophylactic and antirelapse activity against P. cynomolgi in monkeys (Ryley and Peters, Reference Ryley and Peters1970). Compound 25 was abandoned after rapid acquisition of resistance was observed in a murine model. Dihydroacridinedione 26 (WR 243,251), discovered in the 1990s, showed potent blood schizonticidal activity in a P. falciparum-infected monkey model (Kesten et al. Reference Kesten, Degnan, Hung, McNamara, Ortwine, Uhlendorf and Werbel1992; Berman et al. Reference Berman, Brown, Miller, Andersen, McGreevy, Schuster, Ellis, Ager and Rossan1994). Mechanistic studies suggested that compound 26 targeted the parasite's mitochondrial respiratory pathway, and modest cross-resistance was noted with atovaquone. Winter et al. have developed a series of ‘endochin like quinolones’ (ELQ) that exhibit potent antimalarial activity (see Fig. 12, Winter et al. 2011). These compounds have been modified to probe optimum substitutions to improve potency and to improve metabolic stability, lacking in the original endochin. The compound ELQ121 possessed very good activity but had solubility issues for oral administration. The polyethylene glycol prodrug, ELQ125, exhibits superior activity in oral administration in a murine malaria model while retaining activity. Most recently, a quinolone, ELQ-300 (Fig. 13), that has good oral bioavailability at therapeutic doses, is metabolically stable, and is highly active in blocking transmission in rodent malaria models has been described (Nilsen et al. Reference Nilsen, LaCrue, White, Forquer, Cross, Marfurt, Mather, Delves, Shackleford, Saenz, Morrisey, Steuten, Mutka, Li, Wirjanata, Ryan, Duffy, Kelly, Sebayang, Zeeman, Noviyanti, Sinden, Kocken, Price, Avery, Angulo-Barturen, Jimenez-Diaz, Ferrer, Herreros, Sanz, Gamo, Bathurst, Burrows, Siegl, Guy, Winter, Vaidya, Charman, Kyle, Manetsch and Riscoe2013). Because of ELQ-300's highly beneficial metabolic profile and activity against both blood and liver stages of the parasite, it is being taken developed as a potential preclinical drug candidate. Zhang et al. have reported a series of studies, based around a quinolone template, aimed at providing inhibitors of cytochrome bc 1 with improved oral bioavailability (Zhang et al. Reference Zhang, Clark, Connelly, Zhu, Min, Guiguemde, Pradhan, Iyer, Furimsky, Gow, Parman, El Mazouni, Phillips, Kyle, Mirsalis and Kiplin Guy2012). From this group's earlier studies, quinolone 27 (Fig. 13) the original lead, exhibited an EC50 in the sub-micromolar range against both K1 (0·3 μ m) and TM90-C2B (0·91 μ m). These investigations also revealed that a meta-substituted aryl group at the 2-position appeared to be an essential feature of potent 4(1H)-quinolones. More recent reports have described the optimization of substitutions of the 4(1H)-quinolones with respect to the aromatic ring at the 2-position and the benzenoid ring.
Fig. 12. Structures of some 4(1H)-quinolone compounds with antiplasmodial activity and the preclinical candidate ELQ-300.
Fig. 13. Structure of intial quinolone lead and SAR-developed improved analogues.
The substitution pattern of the benzenoid ring (compound 27, Fig. 13) was another feature important for antimalarial potency. In a series of quinolone derivatives, the potency against the K1 strain of P. falciparum followed the trend of 7-MeO>7-MeS>7-OH∼7-iPrO>7-Cl≫7-CF3O>7-CF3. This finding demonstrates that a small hydrophobic electron-donating group at the benzenoid 7-position benefits the antimalarial potency of 4(1H)-quinolone analogues, while an electron-withdrawing group causes a significant decrease in potency.
Further studies examining the effect of substitution at the quinolone 3-position provided a series of structure-activity relationships that impart potency. It was found that when (1) a hydrophobic group is presented at the meta position of 2-phenyl ring, (2) 6-halo and 7-methoxy groups were substituted on the benzenoid ring, and (3) an ethyl ester group was maintained at the 3-position, compounds exhibited the best antimalarial activity. Although increasing hydrophobicity with functionalities on the aromatic group at the 2-position improved activity it also reduced aqueous solubility. Two analogues, 28 and 29 (Fig. 13), were identified with good liver microsome stability and oral systemic exposure in mice. Subsequent murine efficacy testing revealed that 28 had suppression activity at 30 mg kg−1 dosage, equivalent to amodiaquine.
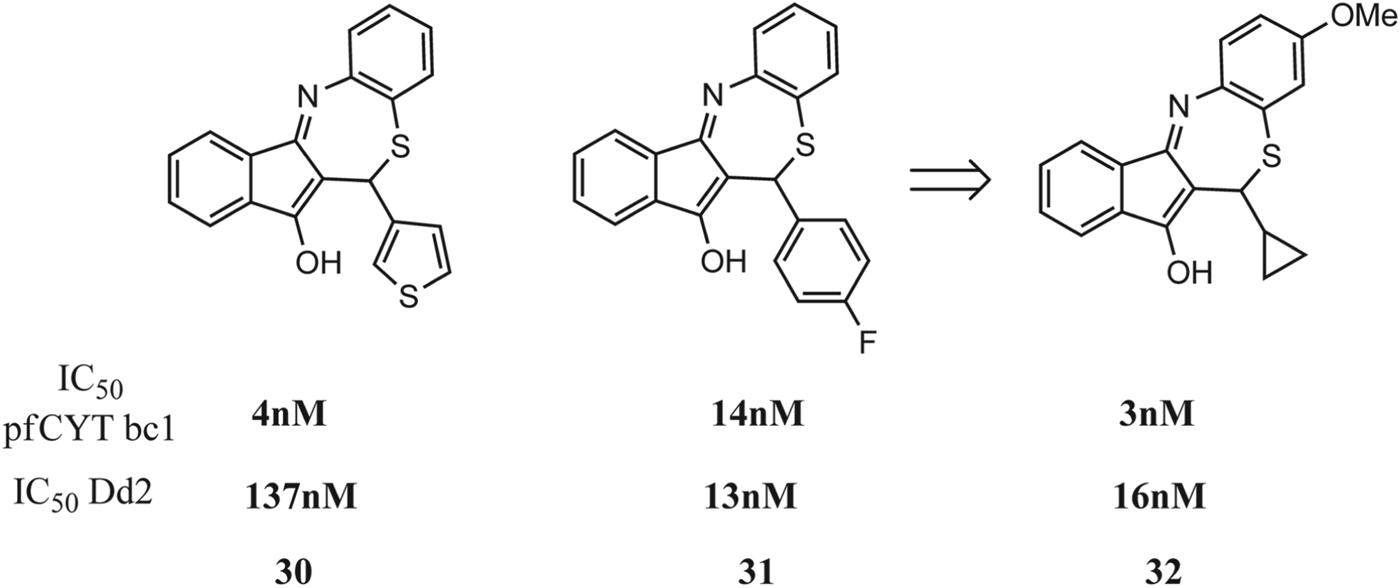
Another recent study, by Dong et al., describes the discovery and development of tetracyclic benzothiazepines (BTZs) as potent and selective antimalarials that target the P. falciparum cytochrome bc 1 complex as the primary functional target (Dong et al. Reference Dong, Urgaonkar, Cortese, Gamo, Garcia-Bustos, Lafuente, Patel, Ross, Coleman, Derbyshire, Clish, Serrano, Cromwell, Barker, Dvorin, Duraisingh, Wirth, Clardy and Mazitschek2011). Investigation of the structure-activity relationship within this previously unexplored chemical scaffold has yielded inhibitors with low nanomolar activity. The authors utilized a large HTS programme to identify novel compounds with antimalarial activity followed by a subsequent screen tailored to target the P. falciparum ETC, two molecules (30 and 31, Fig. 14) with a common tetracyclic benzothiazepine (BTZ) molecular scaffold were identified as having significantly reduced activity against a transgenic scDHOD expressing Dd2 strain with respect to the parental Dd2 strain suggesting that the BTZs target enzymes in the mitochondrial ETC.
Fig. 14. Benzothiazopines with antimalarial activity against plasmodia cytochrome bc1.
A focused library of BTZ derivatives was synthesized for SAR studies to identify analogues with increased potency, and compound 32 was identified as a potent inhibitor of parasite growth with chloroquine-like potency against wild-type 3D7 strain (IC50 = 16 nm), and more importantly, activity against the multi-drug resistant Dd2 strain (IC50 = 16 nm). A clear SAR trend emerged from the analogues where the BTZ benzene ring site was well tolerated for small hydrophobic substituents like methoxy and methyl. In addition, it was observed that non-polar aliphatic groups enhanced antimalarial activity whereas polar substituents diminished potency. Interestingly, minimal cross-resistance exhibited by BTZs with an AVQ-resistant strain indicates that, whereas these molecules likely target the same enzyme, the binding sites are separate and spatially distinct. Although AVQ is a well-studied inhibitor of cytochrome bc 1, the identification of additional inhibitors of this enzyme that do not exhibit cross-resistance with AVQ-resistant parasite strains suggests that the mitochondrial cyt bc 1 still stands as an attractive target for exploitation in new antimalarial development.
P. falciparum type II NADH inhibitors
The cyt bc 1 complex requires reducing equivalents provided by ubiquinol which, in turn, is generated by membrane-bound dehydrogenases upstream in the ETC that catalyse redox reactions by reducing ubiquinone. The parasite lacks the canonical protonmotive NADH dehydrogenase (Complex I) but instead harbours a bacterial-like type II NADH:ubiquinone oxidoreductase, P. falciparum NDH2 (PfNDH2) (Fisher et al. Reference Fisher, Bray, Ward and Biagini2007). PfNDH2 is a single subunit (52 kDa) enzyme involved in the redox reaction of NADH oxidation with subsequent quinol production (Biagini et al. Reference Biagini, Viriyavejakul, O'Neill, Bray and Ward2006). Localized in the mitochondrion, PfNDH2 is a principal electron donor to the ETC, linking fermentative metabolism to the generation of mitochondrial electrochemical membrane potential (Δψm), an essential function for parasite viability (Fig. 1) (Biagini et al. Reference Biagini, Viriyavejakul, O'Neill, Bray and Ward2006).
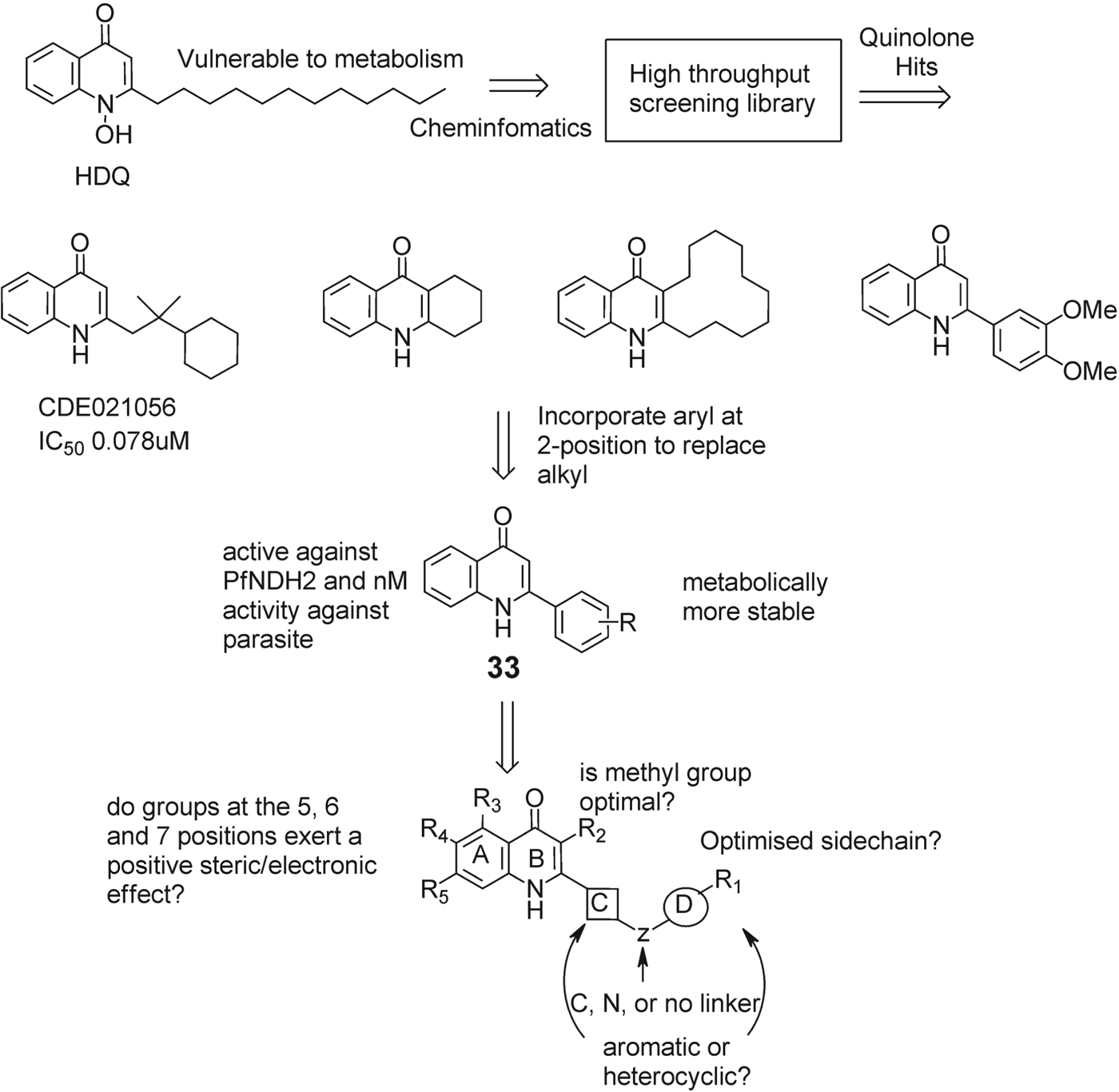
Based on these key observations, Pidathala et al. undertook a medicinal chemistry programme to develop cost effective inhibitors capable of selectively inhibiting PfNDH2 with the goal of providing antimalarials that overcome the limitations of atovaquone (Pidathala et al. Reference Pidathala, Amewu, Pacorel, Nixon, Gibbons, Hong, Leung, Berry, Sharma, Stocks, Srivastava, Shone, Charoensutthivarakul, Taylor, Berger, Mbekeani, Hill, Fisher, Warman, Biagini, Ward and O'Neill2012). In a comprehensive study to identify new molecules, O'Neill et al. employed a range of ligand-based chemoinformatics methods in the rational selection of compounds that were predicted to possess activity against PfNDH2. The chemoinformatics approach were initiated from the identity of only one inhibitor of the target, hydroxy-2-dodecyl-4-(1H)-quinolone (HDQ, Fig. 15) and used molecular fingerprints (Durant et al. Reference Durant, Leland, Henry and Nourse2002), turbosimilarity (Willett, Reference Willett2006), principal components analysis, Bayesian modelling (Geppert et al. Reference Geppert, Vogt and Bajorath2010), and bioisosteric (Liu et al. Reference Liu, Feng, Young and Power2005) replacement in order to select compounds for high-throughput screening (HTS). The compounds were predicted to possess favourable ADMET characteristics (Lipinski Reference Lipinski2000) and the selected compounds were subject to a sequential high-throughput screening methodology using an in vitro assay against recombinant PfNDH2 (Fisher et al. Reference Fisher, Warman, Ward and Biagini2009).
Fig. 15. Development of quinolone template from HTS sampling of commercial library.
Through this process over 40 compounds with IC50 values ranging from below 50 nm to 40 μ m were found but only two of the compounds were selected by more than one chemoinformatic method, justifying the use of several virtual screening approaches. Although several distinct chemotypes were identified from the hit compounds initial investigations led to the selection of the quinolone core as one of the main target chemotypes for SAR development due to its HDQ-like structure.
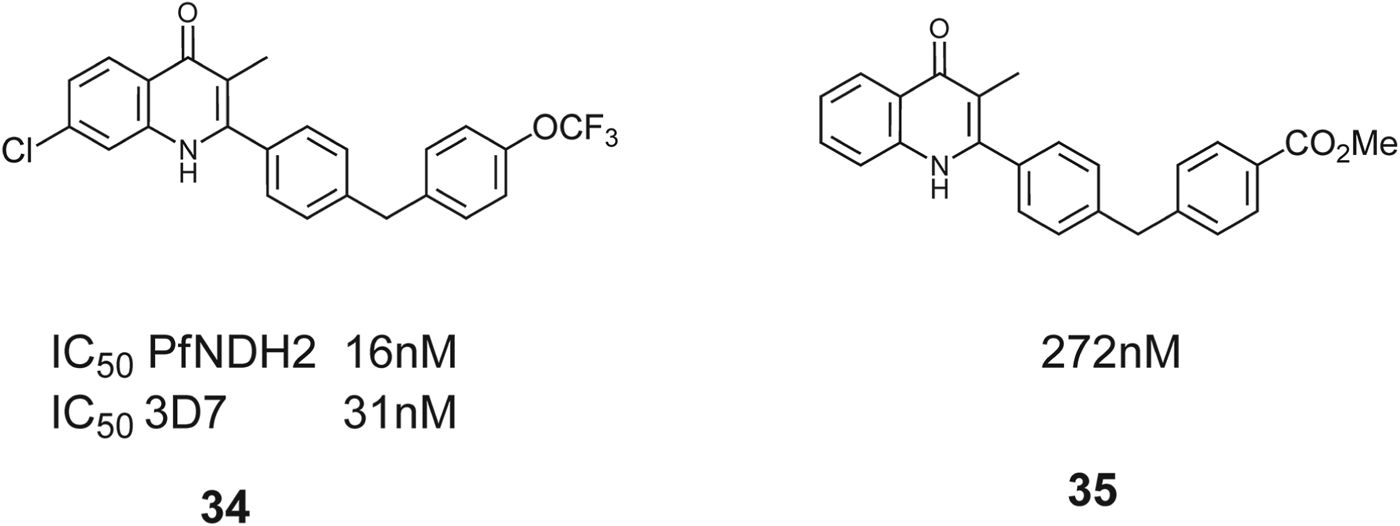
The high potency of hit CDE021056, vs PfNDH2, led to selection of 2-substituted mono-aryl quinolones (33) as a core template with potential for SAR development. The rationale for selection of 2-aryl quinolones was to introduce additional lipophilicity in a region where HDQ contains the flexible aliphatic side-chain. Subsequently, further extension of the side-chain was performed, to impart metabolic stability within the series, and this approach led to eventual identification of early lead compounds for this series. The nature of the group at the 3-position, the electronic/steric effect of substituents placed at the 5, 6 and 7 positions of the quinolone ring, the presence of a nitrogen in the A ring of the quinolone core, and changing from NH to NOH (as in HDQ) were all examined for their effects on activity, stability and toxicity (Biagini et al. Reference Biagini, Fisher, Shone, Mubaraki, Srivastava, Hill, Antoine, Warman, Davies, Pidathala, Amewu, Leung, Sharma, Gibbons, Hong, Pacorel, Lawrenson, Charoensutthivarakul, Taylor, Berger, Mbekeani, Stocks, Nixon, Chadwick, Hemingway, Delves, Sinden, Zeeman, Kocken, Berry, O'Neill and Ward2012; Leung et al. Reference Leung, Gibbons, Amewu, Nixon, Pidathala, Hong, Pacorel, Berry, Sharma, Stocks, Srivastava, Shone, Charoensutthivarakul, Taylor, Berger, Mbekeani, Hill, Fisher, Warman, Biagini, Ward and O'Neill2012). Several compounds proved to be selectively active against the PfNDH2 enzyme. Lead compounds within this series showed antimalarial activity against the 3D7 strain of P. falciparum and PfNDH2 activity in the low nanomolar region and for the most selective quinolone, 34, Fig. 16, a PfNDH2/PfCytbc 1 selectivity ratio of up to 600-fold. It is also observed that other quinolones in this study had the ability to inhibit both PfNDH2 and cyt bc 1 in the low nanomolar range and the authors suggest this dual targeting of two key mitochondrial enzyme targets may prove to be an advantage over single-targeting inhibitors with respect to drug efficacy and delaying the onset of parasite drug resistance. Large electron-withdrawing groups are less well tolerated as seen with 35 (272 nm). As there are several examples of naturally occurring 1-hydroxy-4(1H)-quinolones that are known inhibitors of respiratory and photosynthetic ETCs, it was logical to explore the effect of an N−OH variant of this template on antimalarial activity and PfNDH2 activity, but the general trend pointed to a reduction in activity.
Fig. 16. Example of early lead quinolone with superior antimalarial activity and enzyme selectivity.
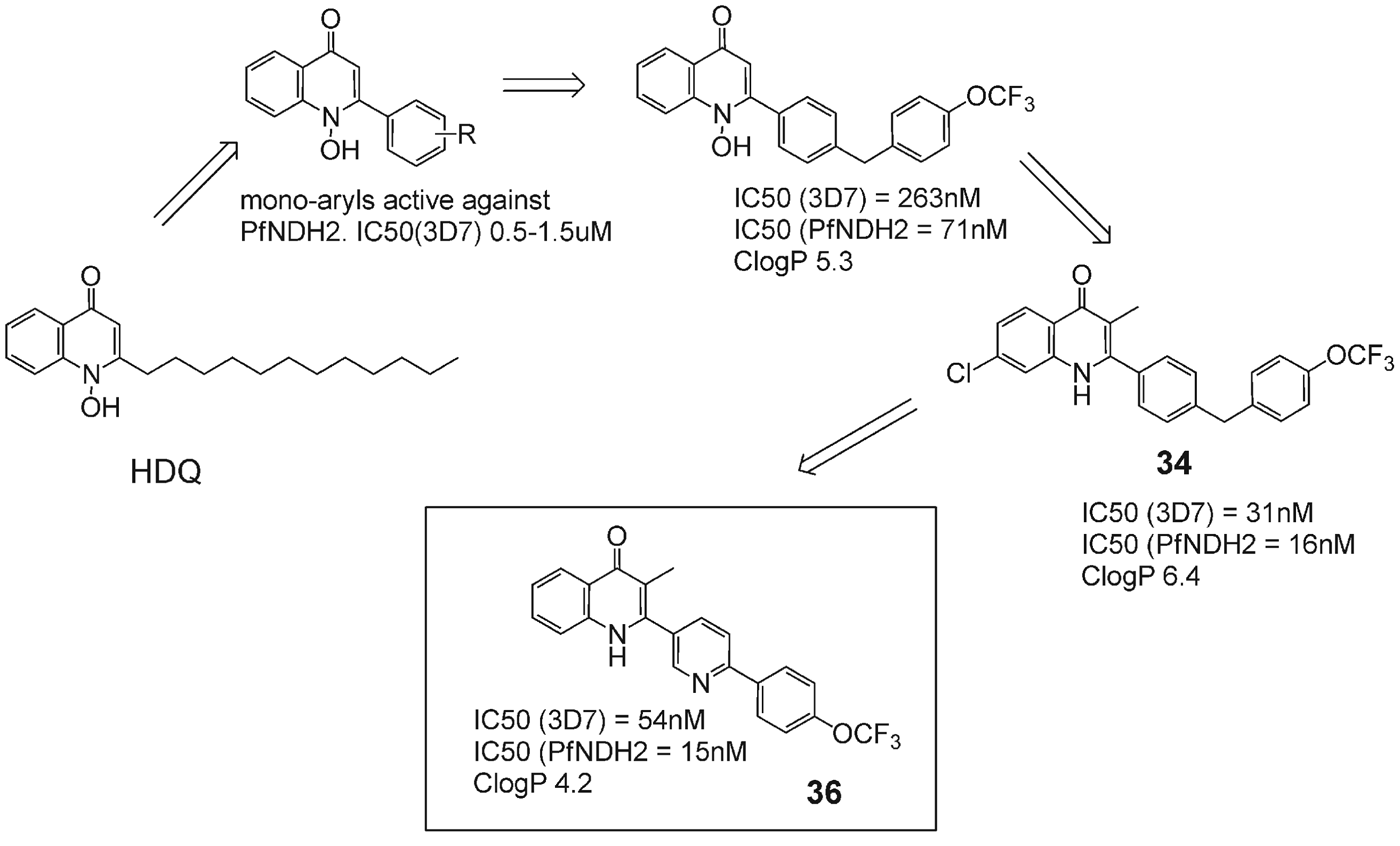
To confirm disruption of pyrimidine biosynthesis by 34, a targeted metabolomics study was undertaken. Metabolites were monitored for response to 34 and atovaquone in both 3D7 and transgenic 3D7-yDHODH·GFP falciparum parasites. The metabolome dynamics following drug exposure confirmed previous reports that atovaquone disrupts pyrimidine metabolism at the point of DHODH as indicated by the accumulation of dihydroorotate and reduction of downstream intermediates. Significantly, no change in pyrimidine metabolite dynamics was observed on atovaquone addition in the transgenic 3D7-yDHODH·GFP strain, confirming the hypothesis that in this strain the yeast DHODH rescues the parasite from cyt bc 1-mediated inhibition of pyrimidine biosynthesis (Biagini et al. Reference Biagini, Fisher, Shone, Mubaraki, Srivastava, Hill, Antoine, Warman, Davies, Pidathala, Amewu, Leung, Sharma, Gibbons, Hong, Pacorel, Lawrenson, Charoensutthivarakul, Taylor, Berger, Mbekeani, Stocks, Nixon, Chadwick, Hemingway, Delves, Sinden, Zeeman, Kocken, Berry, O'Neill and Ward2012). 34 was also shown to disrupt pyrimidine biosynthesis, causing an increase in dihydroorotate and carbonyl-L-aspartate indicative of a similar choke-point at DHODH (Fig. 15B and C). These data indicated that 34 caused parasite death by inhibiting pyrimidine biosynthesis by the collapse of mitochondrial function. From dosing and formulation problems encountered in preliminary animal studies the authors concluded that Clog P needed to be reduced and aqueous solubility needed to be enhanced in order to administer the drug in a suitable vehicle without the need for a pro-drug approach.
Investigations into possible solutions to reduce ClogP revealed that the incorporation of a heterocycle into the side-chain was imperative to achieving this. Introduction of various heterocycles into the quinolone side-chain led to the selection of a series of compounds containing a pyridine group within the side-chain (Leung et al. Reference Leung, Gibbons, Amewu, Nixon, Pidathala, Hong, Pacorel, Berry, Sharma, Stocks, Srivastava, Shone, Charoensutthivarakul, Taylor, Berger, Mbekeani, Hill, Fisher, Warman, Biagini, Ward and O'Neill2012). Incorporation of a pyridine group reduces ClogP as in 36, Fig. 17, improves aqueous solubility, and allows the possibility of salt formation because of the presence of the basic nitrogen (pKa = 4·7).
Fig. 17. Development of late lead quinolone 35 with improved potency and physicochemical profile.
In vivo experiments using P. berghei-infected mice demonstrated that 34 when given orally at 20 mg kg−1 resulted in complete parasite clearance in the Peters’ standard 4-day suppressive test. Compound 36 has been selected as a lead template for further preclinical investigation in the murine model of malaria.
CONCLUSION
The Plasmodium ETC is a viable target for antimalarial drug development. As described, several enzymes along the mitochondrial pathway have been exploited as choke-points to bring about parasite death effectively, and other potential ETC components await chemical and genetic validation. The challenge that remains is to bring the current cluster of drug leads through preclinical evaluation and medicinal chemistry development through phased clinical trials in humans and ultimately field deployment. Currently a number of compounds are at crucial preclinical stages in their development and their ultimate success should be determined within the next few years.
ACKNOWLEDGEMENTS
The authors would like to thank the Medical Research Council (MRC DPFS), Wellcome Trust Seeding Drug Discovery and the Medicines for Malaria Venture (MMV) for support of medicinal chemistry research in this area.