INTRODUCTION
Malaria
Malaria is a disease caused by infection of a human host with protozoan parasites of the genus Plasmodium, and is a devastating global health issue with approximately 200 million cases and 1 million deaths in 2010 alone (Murray et al. Reference Murray, Rosenfeld, Lim, Andrews, Foreman, Haring, Fullman, Naghavi, Lozano and Lopez2012). The complex life cycle of malaria parasites spreads across two hosts and five host tissues whilst undergoing at least ten distinct morphological transitions (Sturm et al. Reference Sturm, Amino, van de Sand, Regen, Retzlaff, Rennenberg, Krueger, Pollok, Menard and Heussler2006; Mackinnon and Marsh Reference Mackinnon and Marsh2010). Replication of parasites and subsequent rupture of erythrocytes in the intra-erythrocytic stages are responsible for the clinical symptoms of malaria, and the majority of drugs target these asexual (human-host) stages of the life cycle. Some species of malaria, most notably Plasmodium vivax, can exist in a latent liver hypnozoite form that can cause relapse even after clearance of bloodstream parasites (Derbyshire et al. Reference Derbyshire, Prudêncio, Mota and Clardy2012; Rodrigues et al. Reference Rodrigues, Prudencio, Moreira, Mota and Lopes2012). Of the five relevant species of human parasite, the vast majority of deaths occur from Plasmodium falciparum infections, which is the typical cause of severe malaria (Claessens et al. Reference Claessens, Adams, Ghumra, Lindergard, Buchan, Andisi, Bull, Mok, Gupta, Wang, Turner, Arman, Raza, Bozdech and Rowe2012). This has led to the majority of drug discovery efforts focusing on P. falciparum, typically at the expense of other species. Although the demand for new P. falciparum drugs is in no doubt, P. vivax is responsible for the majority of worldwide malaria endemicity (Price et al. Reference Price, Douglas and Anstey2009; WHO, 2011). However, difficulties culturing the parasite (Udomsangpetch et al. Reference Udomsangpetch, Somsri, Panichakul, Chotivanich, Sirichaisinthop, Yang, Cui and Sattabongkot2007) along with challenges of imaging and targeting the hypnozoite liver stages (Meister et al. Reference Meister, Plouffe, Kuhen, Bonamy, Wu, Barnes, Bopp, Borboa, Bright, Che, Cohen, Dharia, Gagaring, Gettayacamin, Gordon, Groessl, Kato, Lee, McNamara, Fidock, Nagle, Nam, Richmond, Roland, Rottmann, Zhou, Froissard, Glynne, Mazier, Sattabongkot, Schultz, Tuntland, Walker, Zhou, Chatterjee, Diagana and Winzeler2011) have led to a dearth of new P. vivax drugs (Price et al. Reference Price, Douglas and Anstey2009). Medications capable of targeting all relevant species of parasite, and crucially clearing liver-stage parasites, are in great demand.
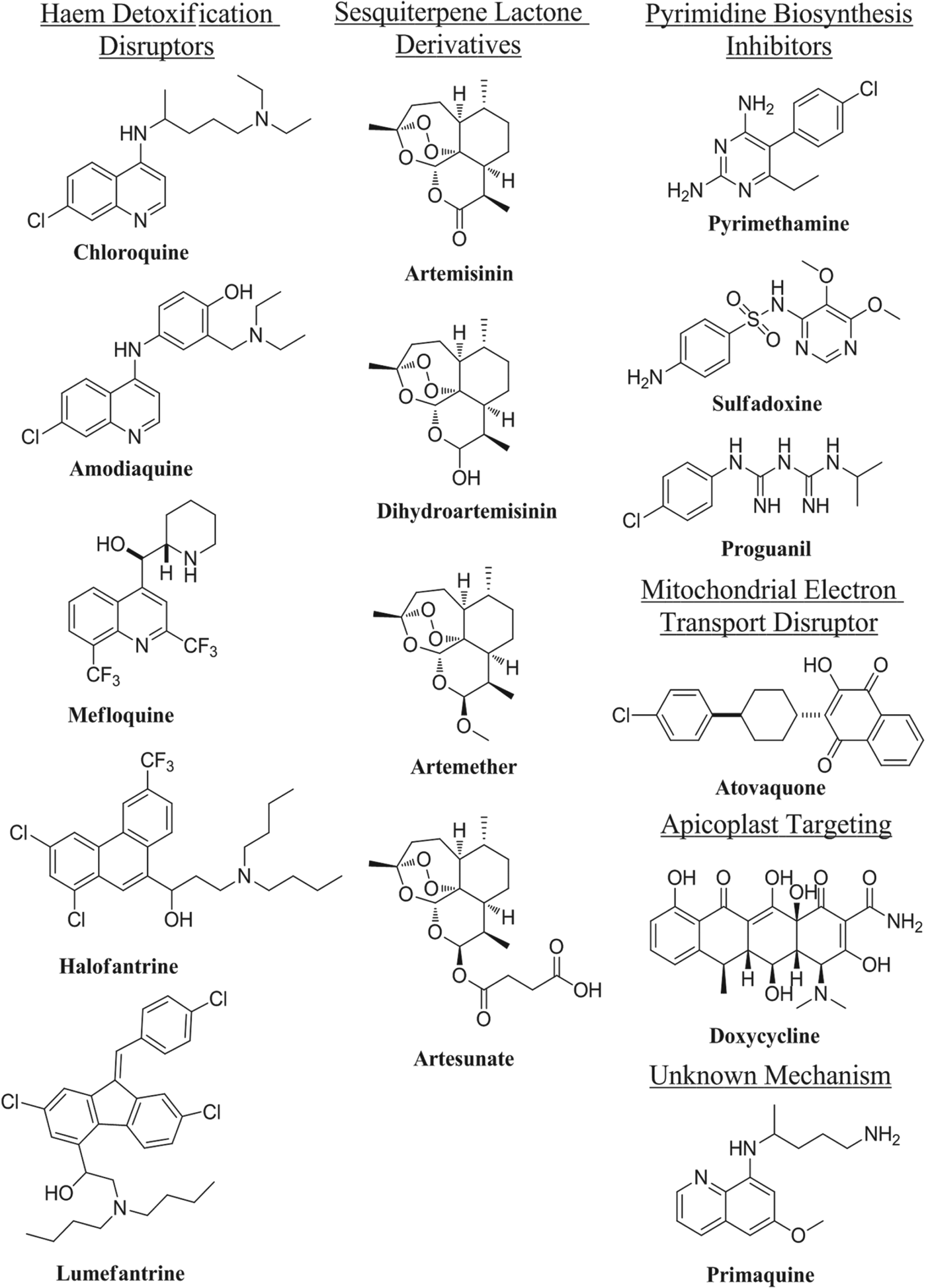
For the latter half of the 20th century, antimalarial drug discovery was a success story for natural product-inspired therapies, by far the most widely used of which are chloroquine (Loeb et al. Reference Loeb, Clark, Coatney, Coggeshall, Dieuaide, Dochez, Hakansson, Marshall, Marvel, McCoy, Sapero, Sebrell, Shannon and Carden1946) and artemisinin (Miller and Su, Reference Miller and Su2011).
Chloroquine was first discovered as a derivative of antimalarial natural product quinine (Krafts et al. Reference Krafts, Hempelmann and Skórska-Stania2012), and has been a first-line antimalarial for over sixty years (Loeb et al. Reference Loeb, Clark, Coatney, Coggeshall, Dieuaide, Dochez, Hakansson, Marshall, Marvel, McCoy, Sapero, Sebrell, Shannon and Carden1946). Studies over the past twenty years have shown that this class of compounds (with the exception of primaquine) is involved in the disruption of haem detoxification by the parasite (Weissbuch and Leiserowitz, Reference Weissbuch and Leiserowitz2008). Artemisinin is a highly effective antimalarial natural product, isolated from Artemisia annua (Miller and Su, Reference Miller and Su2011); its antimalarial action is still under debate, but most hypotheses involve reductive activation of the endoperoxide moiety resulting in parasite death from oxidative damage (O‘Neill and Posner, Reference O'Neill and Posner2004; Li and Zhou, Reference Li and Zhou2010; Slack et al. Reference Slack, Jacobine and Posner2012). Other drug classes used in the treatment of malaria to varying extents include pyrimidine biosynthesis disruptors, drugs that target the apicoplast (Botté et al. Reference Botté, Dubar, McFadden, Maréchal and Biot2012) and drugs discovered by phenotypic screening with unknown targets (e.g. primaquine) (Kappe et al. Reference Kappe, Vaughan, Boddey and Cowman2010). Despite the apparent plethora of antimalarials, drug resistance is a major issue and new medications with distinct mechanisms are constantly required to combat the continued evolution of the parasite (Fidock, Reference Fidock2010; Mackinnon and Marsh, Reference Mackinnon and Marsh2010). This is compounded by the emergence of resistance to the artemisinins in Asia, (Dondorp et al. Reference Dondorp, Nosten, Yi, Das, Phyo, Tarning, Lwin, Ariey, Hanpithakpong, Lee, Ringwald, Silamut, Imwong, Chotivanich, Lim, Herdman, An, Yeung, Singhasivanon, Day, Lindegardh, Socheat and White2009; Phyo et al. Reference Phyo, Nkhoma, Stepniewska, Ashley, Nair, McGready, ler Moo, Al-Saai, Dondorp, Lwin, Singhasivanon, Day, White, Anderson and Nosten2012) reinforcing the urgent requirement for new therapies. Fortunately, a great deal of resource has been directed towards antimalarial drug discovery in the past few decades. Elaboration of the artemisinin pharmacophore has resulted in multiple clinical candidates (Vennerstrom et al. Reference Vennerstrom, Arbe-Barnes, Brun, Charman, Chiu, Chollet, Dong, Dorn, Hunziker, Matile, McIntosh, Padmanilayam, Santo Tomas, Scheurer, Scorneaux, Tang, Urwyler, Wittlin and Charman2004; O'Neill et al. Reference O'Neill, Amewu, Nixon, Bousejra ElGarah, Mungthin, Chadwick, Shone, Vivas, Lander, Barton, Muangnoicharoen, Bray, Davies, Park, Wittlin, Brun, Preschel, Zhang and Ward2010; Charman et al. Reference Charman, Arbe-Barnes, Bathurst, Brun, Campbell, Charman, Chiu, Chollet, Craft, Creek, Dong, Matile, Maurer, Morizzi, Nguyen, Papastogiannidis, Scheurer, Shackleford, Sriraghavan, Stingelin, Tang, Urwyler, Wang, White, Wittlin, Zhou and Vennerstrom2011), although the potential effectiveness of these compounds in artemisinin-resistant regions remains a concern due to the shared mechanism of action. In addition, novel inhibitors of the pyrimidine biosynthetic pathway are in development (Painter et al. Reference Painter, Morrisey, Mather and Vaidya2007) and phenotypic high-throughput screens have resulted in a wealth of information on relevant scaffolds (Plouffe et al. Reference Plouffe, Brinker, McNamara, Henson, Kato, Kuhen, Nagle, Adrián, Matzen, Anderson, Nam, Gray, Chatterjee, Janes, Yan, Trager, Caldwell, Schultz, Zhou and Winzeler2008; Gamo et al. Reference Gamo, Sanz, Vidal, de Cozar, Alvarez, Lavandera, Vanderwall, Green, Kumar, Hasan, Brown, Peishoff, Cardon and Garcia-Bustos2010; Guiguemde et al. Reference Guiguemde, Shelat, Bouck, Duffy, Crowther, Davis, Smithson, Connelly, Clark, Zhu, Jiménez-Díaz, Martinez, Wilson, Tripathi, Gut, Sharlow, Bathurst, Mazouni, Fowble, Forquer, McGinley, Castro, Angulo-Barturen, Ferrer, Rosenthal, DeRisi, Sullivan, Lazo, Roos, Riscoe, Phillips, Rathod, Van Voorhis, Avery and Guy2010), and yielded promising clinical candidates (Rottmann et al. Reference Rottmann, McNamara, Yeung, Lee, Zou, Russell, Seitz, Plouffe, Dharia, Tan, Cohen, Spencer, Gonzalez-Paez, Lakshminarayana, Goh, Suwanarusk, Jegla, Schmitt, Beck, Brun, Nosten, Renia, Dartois, Keller, Fidock, Winzeler and Diagana2010). Among the numerous clinical and preclinical candidates for the treatment for malaria, the vast majority work by existing or unknown mechanisms and are based on known pharmacophores; indeed many are new combinations of existing marketed drugs. Although some have shown efficacy against resistant strains, concerns remain that resistance may develop quickly against already vulnerable mechanisms. New drugs that work by distinct novel biological mechanisms are therefore highly desirable.
The leishmaniases
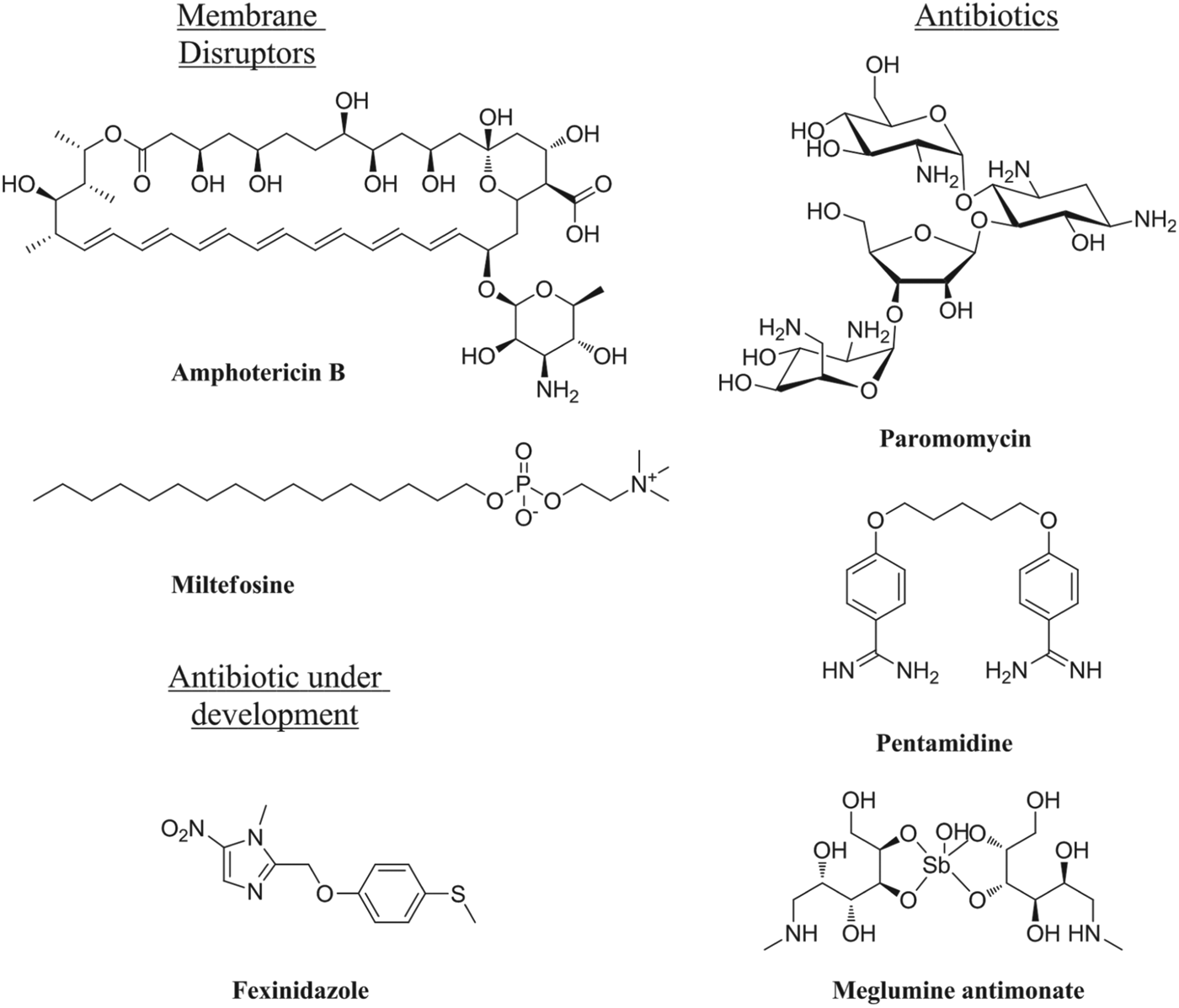
The leishmaniases are the second most prevalent class of parasitic infection after malaria, giving rise to >2 million new cases each year. The disease occurs in three forms, cutaneous (CL), muco-cutaneous (MCL) and the most fatal form, visceral leishmaniasis (VL). The latter is associated with infection by the species Leishmania donovani, while the cutaneous forms are due to infection by multiple species including Leishmania major, L. braziliensis and L. mexicana. The leishmaniases are endemic in more than 90 countries around the world, being particularly prevalent in India, East Africa, Bangladesh and Brazil. Additional clinical issues include post-kala-azar dermal leishmaniasis (PKDL), occurring after the apparent drug cure of VL in certain geographical regions (e.g. Sudan) and difficult to cure with pentavalent antimonials. Combination therapies with therapeutic vaccines (Maroof et al. Reference Maroof, Brown, Smith, Hodgkinson, Maxwell, Losch, Fritz, Walden, Lacey, Smith, Aebischer and Kaye2012) or immune-response activating drugs, such as imiquimod (Arevalo et al. Reference Arevalo, Ward, Miller, Meng, Najar, Alvarez, Matlashewski and Alejandro2001), show some promise. In contrast to the wealth of treatments and drugs in development for malaria, the leishmaniases are poorly provided for. None of the currently available drugs (Fig. 2) were discovered by a rational design process for this neglected disease, and suffer from drawbacks including lack of an oral formulation, prolonged treatment times, high cost of treatment, toxicity, teratogenicity and/or increasing drug resistance. In addition, all work through unknown mechanisms, by disruption of cell membranes or through unspecific antibiotic effects. There are very few drugs in development for these conditions with the most advanced being another antibiotic, fexinidazole (Winkelmann and Raether, Reference Winkelmann and Raether1978), discovered as part of a repurposing initiative (Wyllie et al. Reference Wyllie, Patterson, Stojanovski, Simeons, Norval, Kime, Read and Fairlamb2012).

Fig.1. Structures of clinically-relevant antimalarial drugs.
Fig. 2. Structures of clinically-relevant antileishmanial drugs.
PROTEIN N-MYRISTOYLTRANSFERASE
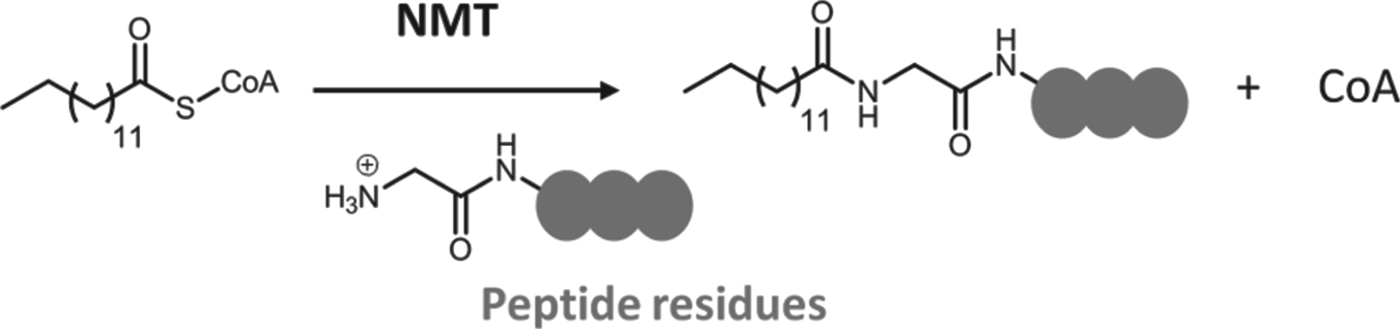
The post-translational modification (PTM) of proteins contributes hugely to the chemical and functional diversity of the cellular proteome and results in the incorporation of molecular motifs not directly encoded by the genome. Protein N-myristoylation is the attachment of a 14-carbon saturated fatty acid, myristate, to the N-terminal glycine residue in a specific set of cellular proteins, catalysed by the enzyme myristoyl CoA:Protein N-myristoyltransferase, NMT (Fig. 3) (Wright et al. Reference Wright, Heal, Mann and Tate2010). Whilst N-myristoylation is often referred to as a PTM, it most commonly occurs co-translationally. Post-translational myristoylation is less well documented but is known to occur following exposure of an internal glycine after cleavage of proteins by caspases during the apoptotic cascade (Zha et al. Reference Zha, Weiler, Oh, Wei and Korsmeyer2000). N-Myristoylation can be involved in protein stability, protein-protein interaction interfaces and association of proteins with membranes.
Fig. 3. The transfer of myristate from myristoyl-CoA to the N-terminal glycine residue of a target peptide (grey circles) by the enzyme N-myristoyltransferase (NMT).
NMT appears to be ubiquitous in eukaryotes, including fungi (Towler et al. Reference Towler, Adams, Eubanks, Towery, Jackson-Machelski, Glaser and Gordon1987; Lodge et al. Reference Lodge, Johnson, Weinberg and Gordon1994; Shaw et al. Reference Shaw, Momany and Momany2002), insects (Ntwasa et al. Reference Ntwasa, Egerton and Gay1997), plants (Boisson et al. Reference Boisson, Giglione and Meinnel2003), mammals (including mouse, rat, cow and human) and the parasitic protozoa P. falciparum (Gunaratne et al. Reference Gunaratne, Sajid, Ling, Tripathi, Pachebat and Holder2000), L. major (Price et al. Reference Price, Menon, Panethymitaki, Goulding, McKean and Smith2003), L. donovani (Brannigan et al. Reference Brannigan, Smith, Yu, Brzozowski, Hodgkinson, Maroof, Price, Meier, Leatherbarrow, Tate, Smith and Wilkinson2010) and Trypanosoma brucei (Price et al. Reference Price, Menon, Panethymitaki, Goulding, McKean and Smith2003). NMT has been shown to be essential for survival in the bloodstream form of T. brucei (Price et al. Reference Price, Menon, Panethymitaki, Goulding, McKean and Smith2003, Reference Price, Guther, Ferguson and Smith2010), in insect stages of L. major (Price et al. Reference Price, Menon, Panethymitaki, Goulding, McKean and Smith2003) and L. donovani (Brannigan et al. Reference Brannigan, Smith, Yu, Brzozowski, Hodgkinson, Maroof, Price, Meier, Leatherbarrow, Tate, Smith and Wilkinson2010) and most recently in the rodent malaria parasite P. berghei (Pino et al. Reference Pino, Sebastian, Kim, Bush, Brochet, Volkmann, Kozlowski, Llinás, Billker and Soldati-Favre2012).
NMT structure and mechanism
The enzyme catalytic cycle has been well-studied in yeast, and follows a Bi-Bi mechanism (Towler et al. Reference Towler, Adams, Eubanks, Towery, Jackson-Machelski, Glaser and Gordon1987; Rudnick et al. Reference Rudnick, McWherter, Rocque, Lennon, Getman and Gordon1991). Myristoyl-CoA (Myr-CoA) binds to the apo-enzyme, inducing a conformational change that allows the protein substrate to bind. Myristate is transferred by attack of the N-terminal glycine amine of the peptide on the thioester carbonyl of Myr-CoA (Rudnick et al. Reference Rudnick, McWherter, Rocque, Lennon, Getman and Gordon1991); CoA is released followed by the myristoylated substrate. The first crystal structure of an NMT to be published was Candida albicans NMT (CaNMT) (Weston et al. Reference Weston, Camble, Colls, Rosenbrock, Taylor, Egerton, Tucker, Tunnicliffe, Mistry, Mancia, de la Fortelle, Irwin, Bricogne and Pauptit1998). Crystal structures of Saccharomyces cerevisiae (ScNMT)(Bhatnagar et al. Reference Bhatnagar, Futterer, Farazi, Korolev, Murray, Jackson-Machelski, Gokel, Gordon and Waksman1998; Farazi et al. Reference Farazi, Waksman and Gordon2001b), and more recently L. donovani NMT (LdNMT) (Brannigan et al. Reference Brannigan, Smith, Yu, Brzozowski, Hodgkinson, Maroof, Price, Meier, Leatherbarrow, Tate, Smith and Wilkinson2010), L. major (LmNMT) (Frearson et al. Reference Frearson, Brand, McElroy, Cleghorn, Smid, Stojanovski, Price, Guther, Torrie, Robinson, Hallyburton, Mpamhanga, Brannigan, Wilkinson, Hodgkinson, Hui, Qiu, Raimi, van Aalten, Brenk, Gilbert, Read, Fairlamb, Ferguson, Smith and Wyatt2010), and the P. vivax enzyme (Goncalves et al. Reference Goncalves, Brannigan, Whalley, Ansell, Saxty, Holder, Wilkinson, Tate and Leatherbarrow2012b), followed. These structures and others provide insight into the binding sites of Myr-CoA, peptide substrates and inhibitors. Published structures are consistent with a highly conserved Myr-CoA binding mode, with Myr-CoA binding in a bent ‘question mark’ conformation (Fig. 4B). The thioester carbonyl is placed into an ‘oxyanion hole’, which activates it for nucleophilic attack, and the fatty acyl chain of Myr-CoA inserts into a deep, hydrophobic pocket. The peptide N-terminal glycine ammonium interacts electrostatically with the buried carboxylate of the C-terminal enzyme residue (Farazi et al. Reference Farazi, Waksman and Gordon2001b), which is responsible for deprotonation of the ammonium so that the generated nucleophilic amine can attack the Myr-CoA thioester (Farazi et al. Reference Farazi, Manchester, Waksman and Gordon2001a, Reference Farazi, Waksman and Gordonb).
Fig. 4. (A) Ternary co-crystal structure of ScNMT (grey surface) with bound peptide substrate (green) and a non-hydrolysable myristoyl-CoA analogue (cyan) bound in the active site. PDB ID: 1IID, Farazi et al. (Reference Farazi, Waksman and Gordon2001b). (B) Myr-CoA or NHM from co-crystal structures with different NMTs; 4A95: NHM in PvNMT, green; 2WUU: NHM in LdNMT, cyan; 2P6E: Myr-CoA in ScNMT, yellow; 1IIC: Myr-CoA in ScNMT, purple; 3IWE: Myr-CoA in HsNMT1, orange; 1IYK: Myr-CoA in CaNMT, blue. Images generated using PyMOL (DeLano Scientific).
NMT substrate specificity
NMT appears to be highly specific for transfer of C14 fatty acids, tolerating only slight changes to chain length (reviewed in Wright et al. Reference Wright, Heal, Mann and Tate2010). However, peptide substrate specificity is complex and there is no definitive ‘myristoylation motif’, beyond the requirement for an N-terminal glycine. This requirement may be mechanism-based: Gordon and co-workers have suggested that rotation of the peptide N-terminal amine about the peptide backbone aligns it for attack on the thioester, and that such a rotation may be hindered for residues with β-substituents, i.e. any amino acid except for glycine (Farazi et al. Reference Farazi, Waksman and Gordon2001b). Maurer-Stroh and co-workers used crystal structures and biochemical data to develop a myristoylation predictive tool: the MYR predictor (Maurer-Stroh et al. Reference Maurer-Stroh, Eisenhaber and Eisenhaber2002a, Reference Maurer-Stroh, Eisenhaber and Eisenhaberb), which suggested that as many as 17 residues may be involved in substrate recognition by NMT. A second tool for predicting N-myristoylation, the ‘Myristoylator’, is based upon a different model for prediction (neural networks, or machine learning) but generates similar error rates compared to the MYR predictor (Bologna et al. Reference Bologna, Yvon, Duvaud and Veuthey2004). Both predictors are necessarily based on the set of proteins annotated or predicted to be N-myristoylated using sequence similarity in SwissProt. Unlike the acyl-CoA binding site, the peptide pocket is not well conserved across NMTs from different species (Maurer-Stroh et al. Reference Maurer-Stroh, Eisenhaber and Eisenhaber2002b). This, together with the observation that NMT is essential for survival, means that this pocket is a target for selective NMT inhibitors (Georgopapadakou Reference Georgopapadakou2002; Price et al. Reference Price, Menon, Panethymitaki, Goulding, McKean and Smith2003).
PROTEIN N-MYRISTOYLATION
Myristoylated proteins have been estimated to make up between 0·5 and 3% of eukaryotic cellular proteomes, depending on the species and in silico model used (Maurer-Stroh et al. Reference Maurer-Stroh, Eisenhaber and Eisenhaber2002a; Martinez et al. Reference Martinez, Traverso, Valot, Ferro, Espagne, Ephritikhine, Zivy, Giglione and Meinnel2008). In the majority of myristoylated proteins studied so far, myristate has a role in transient membrane localization. The ‘two-signal’ membrane binding model suggests that strong membrane localization is only achieved when a second feature of the protein complements myristoylation (Resh, Reference Resh1994). This may be a second acyl group near the N-terminus, such as palmitate (which is usually attached to the side-chain of cysteine residues), a polybasic cluster of amino acids that interacts with membrane phospholipid acidic head groups, or a domain that interacts with another membrane-bound protein (Resh, Reference Resh2006a). Membrane binding in some myristoylated proteins is dynamically regulated via so-called ‘myristoyl switches’, as in the ARF-GTPases, where a change in the ligand bound (from GTP to GDP) causes a conformational change that exposes a hydrophobic pocket that binds myristate, sequestering the fatty acyl chain so that it can no longer interact with the membrane bilayer (Goldberg, Reference Goldberg1998). Myristate-mediated membrane binding is clearly an important mechanism and numerous studies have concluded that it often contributes to the function or regulation of the target protein.
Detecting N-myristoylation in protozoan parasites
Predicting and validating the N-myristoylation of potential substrate proteins in general is challenging due to the complex substrate specificity of NMT and difficulties inherent in detecting protein lipidation. Some proteins can be assigned as likely NMT substrates based on homology; an example being the ARF-GTPases, a class of proteins present in all eukaryotes, having common roles and known to be N-myristoylated in many organisms (Donaldson and Jackson, Reference Donaldson and Jackson2011). For protozoan parasite proteins sharing no sequence identity with generic eukaryotic proteins the main recourse is bioinformatic prediction. However, these tools are necessarily based on known N-myristoylated proteins, of which few have been reported in protozoan parasites, and many of these predictions still require experimental proof. Demonstrating N-myristoylation of a protein in its native context is non-trivial, and thus non-native approaches predominate. A candidate protein is often over-expressed as a GFP or other tagged construct, or the protein of interest is co-expressed with NMT in E. coli, and metabolic radiolabelling with myristate or mass spectrometry is used to demonstrate myristoylation. Lipidation of the protein of interest shown by mass spectrometry, radiolabelling or well characterized chemical probes in the wild-type parasite constitute the only methods for direct proof of N-myristoylation in the native context, whereas lipidation of an over-expressed construct is good evidence. Other data, based on co-expression of the protein with NMT in E. coli or the effects of mutagenesis on membrane localization, are merely suggestive of N-myristoylation.
Protein myristoylation in Plasmodium species
Relatively little is known about which proteins are N-myristoylated in Plasmodium species. P. falciparum possesses a single NMT isoform (Gunaratne et al. Reference Gunaratne, Sajid, Ling, Tripathi, Pachebat and Holder2000) which is able to transfer myristate from Myr-CoA to a peptide substrate based on PfARF1 (Gunaratne et al. Reference Gunaratne, Sajid, Ling, Tripathi, Pachebat and Holder2000). Experimentally studied substrates have roles in life cycle regulation or progression (calcium dependent protein kinase 1 [CDPK1] and Calpain) (Moskes et al. Reference Moskes, Burghaus, Wernli, Sauder, Durrenberger and Kappes2004; Russo et al. Reference Russo, Oksman and Goldberg2009a, Reference Russo, Oksman, Vaupel and Goldbergb), host cell invasion (45 kDa glideosome associated protein [GAP45]) (Rees-Channer et al. Reference Rees-Channer, Martin, Green, Bowyer, Grainger, Molloy and Holder2006), trafficking (ARF1) (Leber et al. Reference Leber, Skippen, Fivelman, Bowyer, Cockcroft and Baker2009), Golgi function (GRASP1) (Struck et al. Reference Struck, de Souza Dias, Langer, Marti, Pearce, Cowman and Gilberger2005) and energy metabolism (Adenylate kinase 2, AK2) (Rahlfs et al. Reference Rahlfs, Koncarevic, Iozef, Mailu, Savvides, Schirmer and Becker2009). However, only GAP45 and CDPK1 have been shown to be N-myristoylated in their native context (Moskes et al. Reference Moskes, Burghaus, Wernli, Sauder, Durrenberger and Kappes2004; Rees-Channer et al. Reference Rees-Channer, Martin, Green, Bowyer, Grainger, Molloy and Holder2006), whilst evidence for N-myristoylation of Calpain is based on radiolabelling of a Calpain-GFP construct (Russo et al. Reference Russo, Oksman and Goldberg2009a). For other potential targets such as Armadillo repeats only protein (ARO), AK2 and GRASP1, evidence is limited (Struck et al. Reference Struck, de Souza Dias, Langer, Marti, Pearce, Cowman and Gilberger2005; Rahlfs et al. Reference Rahlfs, Koncarevic, Iozef, Mailu, Savvides, Schirmer and Becker2009; Cabrera et al. Reference Cabrera, Herrmann, Warszta, Santos, John Peter, Kono, Debrouver, Jacobs, Spielmann, Ungermann, Soldati-Favre and Gilberger2012). CDPK1 requires N-myristoylation for membrane localization (Moskes et al. Reference Moskes, Burghaus, Wernli, Sauder, Durrenberger and Kappes2004), and its gene cannot be knocked out in P. falciparum or the rodent parasite P. berghei, implying essentiality (Kato et al. Reference Kato, Sakata, Breton, Le Roch, Nagle, Andersen, Bursulaya, Henson, Johnson, Kumar, Marr, Mason, McNamara, Plouffe, Ramachandran, Spooner, Tuntland, Zhou, Peters, Chatterjee, Schultz, Ward, Gray, Harper and Winzeler2008; Tewari et al. Reference Tewari, Straschil, Bateman, Bohme, Cherevach, Gong, Pain and Billker2010). In addition, CDPK1 has key functions in multiple stages of the parasite life cycle and is involved in translational activation during sexual development (Sebastian et al. Reference Sebastian, Brochet, Collins, Schwach, Jones, Goulding, Rayner, Choudhary and Billker2012). Another potentially myristoylated kinase, CDPK4, has been shown to be essential for sexual reproduction and mosquito transmission in P. berghei (Billker et al. Reference Billker, Dechamps, Tewari, Wenig, Franke-Fayard and Brinkmann2004). GAP45 is an N-myristoylated protein with a direct role in parasite invasion of RBCs. It is localized at the inner membrane complex (IMC), a series of membrane structures lying beneath the parasite plasma membrane (PM) (Jones et al. Reference Jones, Kitson and Rayner2006). An actomyosin motor, located between the IMC and the PM, drives merozoite invasion, allowing the parasite to enter the RBC (Baum et al. Reference Baum, Richard, Healer, Rug, Krnajski, Gilberger, Green, Holder and Cowman2006). In Toxoplasma gondii, GAP45 is essential to the function of the motor, and therefore for host cell egress, motility and invasion: it has a role in the recruitment of the motor complex and there is evidence for a structural role in maintaining pellicle cohesion during invasion, presumably holding the IMC and PM together (Frenal et al. Reference Frenal, Polonais, Marq, Stratmann, Limenitakis and Soldati-Favre2010). Recent data on the localization of PfGAP45 and N- or C-terminal mutants are consistent with this role in spanning the IMC-PM gap (Ridzuan et al. Reference Ridzuan, Moon, Knuepfer, Black, Holder and Green2012).
Protein myristoylation in Leishmania species
As with Plasmodium, very few proteins have been experimentally validated as N-myristoylated in Leishmania species. Known NMT substrates include proteins involved in trafficking (ARL1 (Sahin et al. Reference Sahin, Espiau, Tetaud, Cuvillier, Lartigue, Ambit, Robinson and Merlin2008)) and proteins of unknown function, such as HASPB, a member of a family of hydrophilic acylated surface proteins expressed in the host and required for parasite development in the insect vector (Denny et al. Reference Denny, Gokool, Russell, Field and Smith2000; Sadlova et al. Reference Sadlova, Price, Smith, Votypka, Volf and Smith2010). A family of ‘small myristoylated proteins’ with probable functions at the flagellum (Tull et al. Reference Tull, Vince, Callaghan, Naderer, Spurck, McFadden, Currie, Ferguson, Bacic and McConville2004, Reference Tull, Heng, Gooley, Naderer and McConville2012) and a protein phosphatase (PPEF) (Mills et al. Reference Mills, Price, Johner, Emerson and Smith2007) have also been reported. In the related trypanosomatid parasites T. brucei and T. cruzi, known likely substrates include TbARF1 (Price et al. Reference Price, Stark and Smith2007), TbARL1 (Price et al. Reference Price, Panethymitaki, Goulding and Smith2005), TbARL6 (Price et al. Reference Price, Hodgkinson, Wright, Tate, Smith, Carrington, Stark and Smith2012), cytoskeletal protein TbCAP5.5 (Hertz-Fowler et al. Reference Hertz-Fowler, Ersfeld and Gull2001), which is involved in cell morphogenesis (Olego-Fernandez et al. Reference Olego-Fernandez, Vaughan, Shaw, Gull and Ginger2009), a flagellar-localized protein (TcFCaBP) (Godsel and Engman, Reference Godsel and Engman1999) and a metacaspase implicated in virulence (Proto et al. Reference Proto, Castanys-Munoz, Black, Tetley, Moss, Juliano, Coombs and Mottram2011). N-Myristoylation of native protein has only been demonstrated directly for LdSMP1 (Tull et al. Reference Tull, Vince, Callaghan, Naderer, Spurck, McFadden, Currie, Ferguson, Bacic and McConville2004), TcFCaBP (Godsel and Engman, Reference Godsel and Engman1999), TbCAP5.5 (Hertz-Fowler et al. Reference Hertz-Fowler, Ersfeld and Gull2001) and TbARL6 (Price et al. Reference Price, Hodgkinson, Wright, Tate, Smith, Carrington, Stark and Smith2012), but there is evidence for lipidation of LmPPEF (Mills et al. Reference Mills, Price, Johner, Emerson and Smith2007) and HASPB (Denny et al. Reference Denny, Gokool, Russell, Field and Smith2000) in Leishmania. In all other cases evidence is limited. Myristoylation of LdARL1 is essential for localization to the Golgi (Sahin et al. Reference Sahin, Espiau, Tetaud, Cuvillier, Lartigue, Ambit, Robinson and Merlin2008) and ARL3, which is involved in maintaining the flagellum of promastigotes, is also thought to be myristoylated (Cuvillier et al. Reference Cuvillier, Redon, Antoine, Chardin, DeVos and Merlin2000). A number of other proteins have been shown to carry a dual acylation motif, including HASPB, which localises to the outer leaflet of the PM in infective stages (Denny et al. Reference Denny, Gokool, Russell, Field and Smith2000). N-terminal N-myristoylation and internal S-palmitoylation are both required for this targeting. Similarly, SMP-1 is flagellum-targeted by myristoylation and palmitoylation (Tull et al. Reference Tull, Vince, Callaghan, Naderer, Spurck, McFadden, Currie, Ferguson, Bacic and McConville2004). A bioinformatic approach predicted around 60 N-myristoylated proteins in Leishmania (Mills et al. Reference Mills, Price, Johner, Emerson and Smith2007), many of which are of unknown function and share little identity with other eukaryotic proteins, suggestive of parasite-specific roles.
Current challenges in defining the N-myristoylated parasite proteome
The handful of identified NMT substrates reflects what is known in other organisms: that N-myristoylation is involved in crucial cellular processes. However, a comprehensive understanding of the N-myristome and the roles of N-myristoylated proteins in protozoa, particularly in infective stages, is still lacking. This is partly due to technical limitations associated with these organisms – for example, challenges in the genetic manipulation of P. falciparum and L. donovani intracellular human infective stages, and a lack of functional analysis of many of the target proteins – but also because detecting lipidation of a native protein is inherently difficult. Radiolabelling with [3H]myristate or other fatty acids such as palmitate with detection by fluorography is the most common traditional method for studying protein acylation, but is a laborious process due to the very long (weeks or months) exposure times required. Mass spectrometry is a powerful and continually advancing technique that can be used to detect PTMs such as fatty acylation, but generally the protein of interest must first be highly enriched and lipophilic fatty acylated proteins can be lost during preparation (Resh, Reference Resh2006b). The concentration range of proteins within cells is huge – around 5–6 orders of magnitude (Tyers and Mann, Reference Tyers and Mann2003) – and this further hinders detection of rare proteins in complex mixtures. Chemical proteomic approaches have revolutionized the field of PTM in the past decade, particularly for low abundance PTMs such as N-myristoylation. Acyl biotin exchange chemistry (ABE), where palmitate is exchanged site-specifically for biotin at the site of PTM (Roth et al. Reference Roth, Wan, Green, Yates and Davis2006), is widely used for analysing S-palmitoylation and was recently applied in P. falciparum blood stages with the identification of several thousand new potential targets (Jones et al. Reference Jones, Collins, Goulding, Choudhary and Rayner2012). Another methodology, the bioorthogonal probe approach, involves the metabolic incorporation of a PTM substrate analogue containing a small, biologically inert chemical tag into proteins in live cells; the tag is subsequently functionalized with useful labels for detection or identification (Wright et al. Reference Wright, Heal, Mann and Tate2010; Hang et al. Reference Hang, Wilson and Charron2011). These approaches have been used to great effect for the profiling of acylated proteins in mammalian cells and tagged lipid analogues were recently applied to identify S-palmitoylated proteins in P. falciparum (in parallel with ABE) (Jones et al. Reference Jones, Collins, Goulding, Choudhary and Rayner2012) and to demonstrate N-myristoylation of T. brucei ARL6, a protein with a putative role in flagellum biogenesis (Price et al. Reference Price, Hodgkinson, Wright, Tate, Smith, Carrington, Stark and Smith2012). Chemical probes have the potential to greatly expand the list of N-myristoylated proteins in parasitic protozoa and contribute to our understanding of NMT as a drug target.
TOWARDS NMT INHIBITORS AS ANTIMALARIAL OR ANTILEISHMANIAL AGENTS
Several lines of evidence suggest that NMT is a promising drug target for malaria and leishmaniasis; it is a monomeric enzyme carrying out a specific modification on substrates involved in diverse and essential pathways, it is essential for viability where genetic validation has been possible, and is constitutively expressed. Furthermore it is genetically and chemically validated as a drug target in T. brucei, where small molecule inhibitors have been shown to be effective in animal models (Frearson et al. Reference Frearson, Brand, McElroy, Cleghorn, Smid, Stojanovski, Price, Guther, Torrie, Robinson, Hallyburton, Mpamhanga, Brannigan, Wilkinson, Hodgkinson, Hui, Qiu, Raimi, van Aalten, Brenk, Gilbert, Read, Fairlamb, Ferguson, Smith and Wyatt2010). The wide variety of NMT substrates may limit the potential for resistance to develop against inhibitors targeting the protein binding site, since mutations in this site could inhibit correct myristoylation of substrates.
Initial research within our group focused on the discovery of plasmodial NMT inhibitors as chemical probes and potential therapeutic agents for the treatment of malaria, due to the availability of chemical starting points for this indication. Drug repositioning, the adaptation of an existing drug for a new indication, is often used to bypass the significant cost of clinical trials as the safety/pharmacokinetic data for these compounds has already been established (Sleigh and Barton, Reference Sleigh and Barton2010). A similar approach can be used for hit discovery (the ‘piggy-back approach’) to discover promising hit series without the cost of an HTS campaign (Gelb et al. Reference Gelb, Van Voorhis, Buckner, Yokoyama, Eastman, Carpenter, Panethymitaki, Brown and Smith2003). In the case of NMT, a wealth of information has been collated with the purpose of generating antifungal NMT inhibitors. It was hypothesized that this information could be used as a valuable resource in the generation of parasitic NMT inhibitors, since CaNMT and PfNMT display 38% identity and 65% similarity.
In the process of validation of a radioactive assay for monitoring N-myristoylation, a library of 43 CaNMT inhibitors containing a benzothiazole scaffold (provided by Pfizer) were screened against PfNMT and Homo sapiens NMT1 (HsNMT1) (Bowyer et al. Reference Bowyer, Gunaratne, Grainger, Withers-Martinez, Wickramsinghe, Tate, Leatherbarrow, Brown, Holder and Smith2007). Of these 43 compounds, dose-response curves were generated for 7 of the most promising inhibitors, four of which reduced parasitaemia in vitro. These compounds had weak enzyme affinity and cellular potency, and displayed very high molecular weight and lipophilicity for compounds with this level of activity. Ligand Efficiency (LE) is a commonly-used measure of how tightly a compound binds to a target protein, relative to its overall size (Hopkins et al. Reference Hopkins, Groom and Alex2004; Bembenek et al. Reference Bembenek, Tounge and Reynolds2009); LE>0·35 is considered favourable, as it suggests scope for substantial optimization. However, LE in this series was around 0·22, limiting the potential for future development. A distinct small library of 25 previously described CaNMT and TbNMT inhibitors was also tested against PfNMT in vitro and compound RO-09-4609 emerged as a promising hit compound, with weak but selective PfNMT affinity. This hit was optimized by iterative medicinal chemistry, resulting in a moderate affinity and highly selective compound Example 26 (Yu et al. 2012), representing a 100-fold affinity improvement over the initial hit (Fig. 5A).The binding mode of these compounds was validated in PvNMT (81% sequence identity to PfNMT), confirming the hypothesis that these compounds are competitive with the peptide substrate; however, LE (0·29) remained sub-optimal. A scaffold-hopping approach was then applied to yield 2,3-benzothiophene Example 9 (Rackham et al. Reference Rackham, Brannigan, Moss, Yu, Wilkinson, Holder, Tate and Leatherbarrow2013), resulting in the most ligand efficient inhibitor discovered at this stage of development against PfNMT (Fig. 5A). Crystallography of this inhibitor series bound to PvNMT confirmed that the 2,3-benzothiophene inhibitors occupy a distinct binding mode to the 2,3,4 benzofurans exemplified by Example 26 (Yu et al. 2012), representing a novel inhibitor series for further development (Fig. 5B) (Rackham et al. Reference Rackham, Brannigan, Moss, Yu, Wilkinson, Holder, Tate and Leatherbarrow2013).
Fig. 5. (A) Summary of the PfNMT inhibitor series obtained by a ‘Piggy-Back’ approach from fungal NMT inhibitor RO-09-4609. (B) Binding mode of Example 26 (Yu et al. 2012) bound to the Plasmodium vivax NMT active site. PDB Accession Code: 4B14.
Whilst the ‘piggy-back’ approach provided a highly ligand-efficient series of PfNMT inhibitors, cross-screening the same set of compounds against L. major NMT failed to identify any starting points (Panethymitaki et al. Reference Panethymitaki, Bowyer, Price, Leatherbarrow, Brown and Smith2006), necessitating alternative hit generation strategies. High-throughput screening has been successful in identifying multiple series of NMT inhibitors, including C. albicans (Masubuchi et al. Reference Masubuchi, Kawasaki, Ebiike, Ikeda, Tsujii, Sogabe, Fujii, Sakata, Shiratori, Aoki, Ohtsuka and Shimma2001; Ohtsuka and Aoki, Reference Ohtsuka and Aoki2003), Aspergillus fumigatus (Bowyer et al. Reference Bowyer, Tate, Leatherbarrow, Holder, Smith and Brown2008), Cryptococcus neoformans (Bowyer et al. Reference Bowyer, Tate, Leatherbarrow, Holder, Smith and Brown2008) and T. brucei (Frearson et al. Reference Frearson, Brand, McElroy, Cleghorn, Smid, Stojanovski, Price, Guther, Torrie, Robinson, Hallyburton, Mpamhanga, Brannigan, Wilkinson, Hodgkinson, Hui, Qiu, Raimi, van Aalten, Brenk, Gilbert, Read, Fairlamb, Ferguson, Smith and Wyatt2010). Although compounds from the latter publication have been co-crystallized with L. major NMT and display excellent enzyme affinity, the cellular potency of this series has not been disclosed. Consequently, we initiated a broader screening programme to identify additional selective inhibitors of Plasmodium and Leishmania NMTs. We screened the 150 000 compound Pfizer Global Diverse Representative Set against LdNMT, anticipating that hits would be likely to possess pan-Leishmania NMT activity since the residues involved in the binding pocket are completely conserved (Brannigan et al. Reference Brannigan, Smith, Yu, Brzozowski, Hodgkinson, Maroof, Price, Meier, Leatherbarrow, Tate, Smith and Wilkinson2010). We also screened the same set against PfNMT, and the initial hit set was supplemented by analogues selected from the remainder of the Pfizer file (∼2·5 million compounds at the time of the high-throughput screen) (Bell et al. Reference Bell, Mills, Williams, Brannigan, Wilkinson, Parkinson, Leatherbarrow, Tate, Holder and Smith2012). In addition, a separate library of 60 000 compounds was screened against PvNMT, in collaboration with MRC Technology, in the hope of identifying distinct hit compounds (Goncalves et al. Reference Goncalves, Brannigan, Thinon, Olaleye, Serwa, Lanzarone, Wilkinson, Tate and Leatherbarrow2012a).
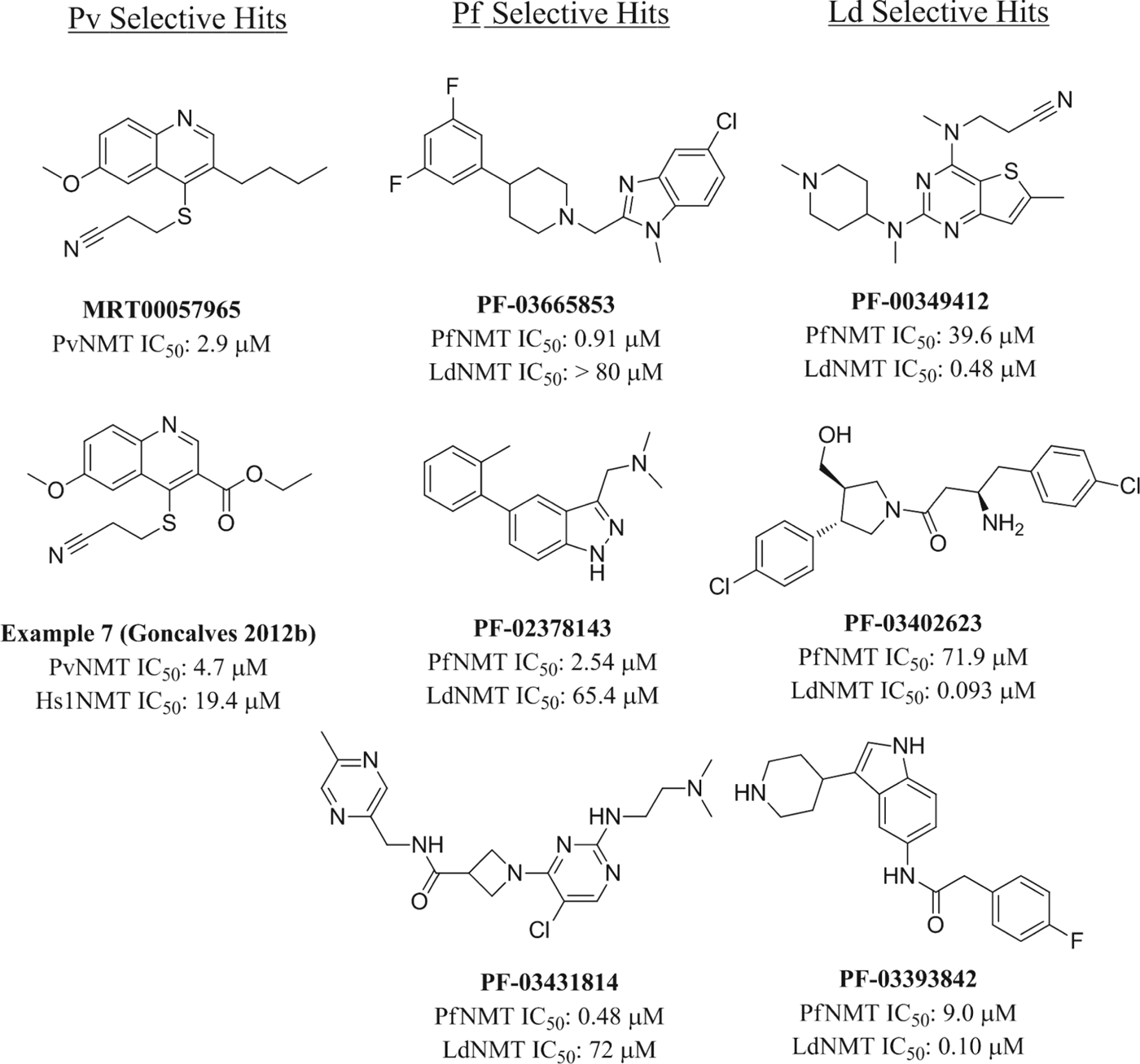
Comparison of the hits from each HTS (Fig. 6) shows a remarkably wide range of structural features. Although many possess a basic centre, a common pharmacophore of several previously described NMT inhibitors, the hits feature primary, secondary and tertiary amines, which may be interacting with the conserved C-terminal Leu of the enzyme. However, the MRCT HTS hit (MRT00057965) is not basic and makes an alternative H-bond interaction with Ser319 (Goncalves et al. Reference Goncalves, Brannigan, Whalley, Ansell, Saxty, Holder, Wilkinson, Tate and Leatherbarrow2012b). Based on the diversity of known binding modes for NMT inhibitors, it is difficult to predict those for novel inhibitor series, and structures of the remaining HTS hits are, as yet, unreported. A limited medicinal chemistry optimization programme based on the MRT hit has been published (Goncalves et al. Reference Goncalves, Brannigan, Whalley, Ansell, Saxty, Holder, Wilkinson, Tate and Leatherbarrow2012b), resulting in a confirmed hit with improved physical properties and moderate selectivity over the human isoforms. In order to address the enzyme selectivity issue, the Pfizer screen hit set was also tested in dose-response assays against both human NMT isoforms and against T. brucei NMT (Bell et al. Reference Bell, Mills, Williams, Brannigan, Wilkinson, Parkinson, Leatherbarrow, Tate, Holder and Smith2012). Consistent with our previous findings, LdNMT seems distinct from the other NMTs in our screening panel, as most hits show excellent selectivity. In contrast, selectivity for PfNMT over either human NMT is more elusive, though the screen did identify two hit series with encouraging profiles.
Fig. 6. Structures of hits obtained by high-throughput screening against protozoal NMTs.
The structural basis for the observed selectivities remains unclear. Analysis of the peptide-binding pocket of LdNMT identified two residue differences with T. brucei NMT (Brannigan et al. Reference Brannigan, Smith, Yu, Brzozowski, Hodgkinson, Maroof, Price, Meier, Leatherbarrow, Tate, Smith and Wilkinson2010), and the same residues are also points of differentiation between LdNMT and both human NMTs. In contrast, the residues lining the binding pocket are conserved between PfNMT and human NMT with the exception of a conservative F334Y change, which is also a point of difference between PfNMT and P. vivax NMT. Plasmodium/human NMT selectivity has been attributed to differences in their ability to tolerate a conformational change in Y296 (HsNMT1 numbering) (Yu et al. 2012). It remains to be seen whether this difference in conformation is observed with other Plasmodium-selective inhibitors.
FUTURE PERSPECTIVES
Two significant hurdles remain for the validation of NMT as a drug target in malaria and leishmaniasis: the challenge of understanding the role of N-myristoylation through understanding of its downstream protein substrates, and proving its essentiality in clinically-relevant parasites.
Protein N-myristoylation is a mechanism used universally by eukaryotes to direct protein localization and hence function, but detecting protein lipidation by traditional methods presents challenges, particularly in P. falciparum and L. donovani which are not easily genetically manipulated and go through distinct life cycle stages in a variety of host environments. Bioinformatic prediction suggests that many proteins with diverse and unknown roles are likely to be myristoylated in parasitic protozoa, but in very few cases has acylation been experimentally validated. The development of new techniques to profile protein lipidation, such as the application of bioorthogonal chemical probes (Heal and Tate, Reference Heal and Tate2010), may allow us to gain a much wider and in-depth understanding of the myristoylated proteome and to explore the downstream effects of NMT inhibition in these important human pathogens.
Demonstration of essentiality and druggability of NMT in P. falciparum and L. donovani requires selective inhibitors, and proof that these compounds act on-target in parasites. Progress towards selective and potent parasite NMT inhibitors is at an exciting stage; Example 9 (Fig. 5) derived from the ‘piggy-back’ approach represents a promising starting point for inhibitor development, demonstrating excellent ligand efficiency and selectivity over the human orthologues. Future development of this series will focus on affinity enhancements whilst maintaining optimum physicochemical properties for drug-like molecules, with the aim of generating a high value lead series for clinical development. Replacement of the alkyl ester in Example 9 with a more biologically stable isostere is a paramount objective for further development since oral administration is a prerequisite for a malaria medication. In addition, the other Plasmodium-selective hits identified (Fig. 6) represent highly promising series for further development, demonstrating a range of chemotypes with varying physicochemical properties. Successful elaboration of any of these hit series into a potent, selective and drug-like inhibitor of NMT would enable investigation in cellular and in vivo models of malaria, providing a chemical tool for the validation of NMT as a drug target in malaria infections. The need for new drugs against leishmaniasis is still more pressing, particularly in view of the relatively neglected nature of this disease. Our recent screening initiative has also opened the door to the development of potent, selective inhibitors of Leishmania NMT, and we anticipate that the available chemical matter will enable both the development of tools to explore the role and importance of NMT in this challenging organism, and new starting points for discovery of antileishmanial drugs.
The historic track record of targeted approaches to treatment of parasitic infections is in general relatively poor, since there are significant barriers to overcome in achieving good translation of enzyme to cellular activity and in vivo efficacy. Future work in our laboratory is focused on overcoming these hurdles to confirm the relevance of NMT inhibition as a valid target for treating parasitic infections.
ACKNOWLEDGEMENTS
The authors gratefully acknowledge members of the Tate, Leatherbarrow, Holder, Smith and Wilkinson labs at Imperial College, MRC National Institute for Medical Research, and the University of York for their advice and assistance with proof reading, and the reviewers for their constructive comments during the preparation of this manuscript.
FINANCIAL SUPPORT
The authors would like to acknowledge the Wellcome Trust (grant no. 087792), the Medical Research Council (grant nos. 0900278 and U117532067), Imperial College, EPSRC and Pfizer for supporting this work.