



INTRODUCTION
Cyclosporiasis, caused by Cyclospora cayetanensis, is an emerging human infection causing gastrointestinal disease around the world (Ortega et al. Reference Ortega, Sterling, Gilman, Cama and Diaz1993; Ortega and Sanchez, Reference Ortega and Sanchez2010; Zhou et al. Reference Zhou, Lv, Wang, Wang, Jian, Zhang, Ning, Fu, Wang, Qi, Yao, Zhao, Zhang, Sun, Shi, Arrowood and Xiao2011; Plutzer and Karanis, Reference Plutzer and Karanis2016). It is also an endemic disease in many developing countries, and is responsible for significant morbidity in children and patients with acquired immunodeficiency syndrome (Ortega and Sanchez, Reference Ortega and Sanchez2010). Since the mid-1990s, numerous sporadic cases or outbreaks of cyclosporiasis have occurred throughout the world, usually associated with individuals returning home from endemic areas or consuming imported contaminated fresh produce (Döller et al. Reference Döller, Dietrich, Filipp, Brockmann, Dreweck, Vonthein, Wagner-Wiening and Wiedenmann2002; Ortega and Sanchez, Reference Ortega and Sanchez2010; Legua and Seas, Reference Legua and Seas2013; Abanyie et al. Reference Abanyie, Harvey, Harris, Wiegand, Gaul, Desvignes-Kendrick, Irvin, Williams, Hall, Herwaldt, Gray, Qvarnstrom, Wise, Cantu, Cantey, Bosch, DA Silva, Fields, Bishop, Wellman, Beal, Wilson, Fiore, Tauxe, Lance, Slutsker and Parise2015; Giangaspero et al. Reference Giangaspero, Marangi, Koehler, Papini, Normanno, Lacasella, Lonigro and Gasser2015).
The clinical diagnosis of cyclosporiasis currently involves the detection of oocysts in the patient's fecal sample with microscopy (Zhou et al. Reference Zhou, Lv, Wang, Wang, Jian, Zhang, Ning, Fu, Wang, Qi, Yao, Zhao, Zhang, Sun, Shi, Arrowood and Xiao2011; Kaminsky et al. Reference Kaminsky, Lagos, Raudales Santos and Urrutia2016), together with a limited number of molecular diagnostic tools, including the analysis of small subunit ribosomal RNA (SSU rRNA) gene (Zhou et al. Reference Zhou, Lv, Wang, Wang, Jian, Zhang, Ning, Fu, Wang, Qi, Yao, Zhao, Zhang, Sun, Shi, Arrowood and Xiao2011) and 70 kDa heat-shock protein (HSP70) gene (Sulaiman et al. Reference Sulaiman, Ortega, Simpson and Kerdahi2014). However, a multilocus sequence typing (MLST) tool for C. cayetanensis based on five microsatellite loci was developed, with high resolution in characterizing C. cayetanensis isolates (Guo et al. Reference Guo, Roellig, Li, Tang, Frace, Ortega, Arrowood, Feng, Qvarnstrom, Wang, Moss, Zhang and Xiao2016). It is also useful for the investigation for case linkage and trace back during disease outbreaks. A population genetic analysis, based on several mini- and microsatellites with sufficient resolution, could also clarify the evolution of C. cayetanensis (Guo et al. Reference Guo, Roellig, Li, Tang, Frace, Ortega, Arrowood, Feng, Qvarnstrom, Wang, Moss, Zhang and Xiao2016). Such genetic structural analyses have also been conducted for Plasmodium falciparum (Tibayrenc and Ayala, Reference Tibayrenc and Ayala2002; Su et al. Reference Su, Hayton and Wellems2007; Buckee and Gupta, Reference Buckee and Gupta2010), Toxoplasma gondii (Gatei et al. Reference Gatei, Das, Dutta, Sen, Cama, Lal and Xiao2007; Khan et al. Reference Khan, Fux, Su, Dubey, Darde, Ajioka, Rosenthal and Sibley2007), Cryptosporidium spp. (Mallon et al. Reference Mallon, MacLeod, Wastling, Smith, Reilly and Tait2003; Tanriverdi et al. Reference Tanriverdi, Markovics, Arslan, Itik, Shkap and Widmer2006; Morrison et al. Reference Morrison, Mallon, Smith, MacLeod, Xiao and Tait2008; Wang et al. Reference Wang, Jian, Zhang, Ning, Liu, Zhao, Feng, Qi, Wang, Lv, Zhao and Xiao2012, Reference Wang, Zhang, Axén, Bjorkman, Jian, Amer, Liu, Feng, Li, Lv, Zhao, Qi, Dong, Wang, Sun, Ning and Xiao2014) and Enterocytozoon bieneusi (Karim et al. Reference Karim, Wang, He, Zhang, Li, Rume, Dong, Qi, Jian, Zhang, Sun, Yang, Zou, Ning and Xiao2014; Li et al. Reference Li, Wan, Yu, Yang, Tao, Jiang and Xiao2016; Wan et al. Reference Wan, Xiao, Zhang, Li, Lu, Song and Li2016).
The examination of C. cayetanensis infections in humans has detected a total of 25 multilocus genotypes (MLGs) in China, the USA, Nepal, Peru and Spain, but there has been no population structure analysis (Guo et al. Reference Guo, Roellig, Li, Tang, Frace, Ortega, Arrowood, Feng, Qvarnstrom, Wang, Moss, Zhang and Xiao2016). To investigate the prevalence of C. cayetanensis in a longitudinal study and to conduct a genetic analysis, patients’ fecal specimens were collected during the seasons of high cyclosporiasis prevalence in Henan Province, central China, in 2011–2015. A population genetic analysis was conducted of these isolates and reference C. cayetanensis isolates from humans.
MATERIALS AND METHODS
Ethics statement
Before the study, the research protocol was reviewed and approved by the Ethics Review Committee of the Henan Center for Disease Control and Prevention, China. Appropriate permission was obtained from the director of the hospitals involved before the stool samples were collected. The attending doctors were notified as soon as possible if any parasite oocysts or cysts were detected to expedite their treatment.
Study area and sampling
This study was conducted in two adjacent urban areas, Zhengzhou (112°42′E–114°14′E, 34°16′N–34°58′N) and Kaifeng (113°52′E–115°15′E, 34°11′N–35°11′N), in central China. A total of 6579 patients (3629 males, 2950 females) at three hospitals (Henan Province People's Hospital, Zhengzhou; Huaihe Hospital, Kaifeng; and 155 Central Hospital, Kaifeng) were enrolled in the study in July and August of 2011–2015. Only data concerning the location, age, sex and presence or absence of diarrhoea were made available to laboratory technicians.
PCR amplification and sequencing
The DNA genome of the parasite in all the specimens was extracted with the E.Z.N.A.R Stool DNA Kit (Omega Biotek Inc., Norcross, Georgia, USA), according to the manufacturer's instructions. The extracted DNAs were stored at −20 °C until analysis.
The C. cayetanensis infections in the patients were screened with the PCR amplification of SSU rRNA gene (Li et al. Reference Li, Xiao, Zhou, Li and Wadeh2007). The positive specimens were further characterized with a PCR analysis of five microsatellite loci (CYC3, CYC13, CYC15, CYC21 and CYC22), as previously described (Guo et al. Reference Guo, Roellig, Li, Tang, Frace, Ortega, Arrowood, Feng, Qvarnstrom, Wang, Moss, Zhang and Xiao2016).
The amplicons of the expected sizes from PCR were sequenced on an ABI PRISM™ 3730 XL DNA Analyzer using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, California, USA). Sequence accuracy was confirmed with bidirectional sequencing and by sequencing a new PCR product if necessary.
MLST analysis
The sequences of all five loci analysed in this study were combined in a single contig, as previously described (Guo et al. Reference Guo, Roellig, Li, Tang, Frace, Ortega, Arrowood, Feng, Qvarnstrom, Wang, Moss, Zhang and Xiao2016), to determine the MLGs, with consideration of both single-nucleotide polymorphisms (SNPs) and short polymorphic insertions or deletions (indels).
The DNA polymorphisms in each of the five markers and in the combined sequences were assessed to calculate the gene diversity (H d) and nucleotide diversity (P i) with DnaSP 5.10.01 (Librado and Rozas, Reference Librado and Rozas2009). Intragenic linkage disequilibrium (LD) was calculated with both two-tailed Fisher's exact test and the χ 2 test to determine the degree of LD. The minimum numbers of recombination events (R m) at each of the five loci and in the concatenated contigs were evaluated. Tests for genetic diversity and neutrality (Fu's F s statistic) were performed at the segregating sites.
The standardized index of association (I A S) and its probability under a null model of complete panmixia were calculated with hypothesis testing using a Monte Carlo simulation with 1000 iterations in software of lian – LInkage ANalysis, version 3·7 (Haubold and Hudson, Reference Haubold and Hudson2000). Values I A S, the distribution of the mismatch values (V D), the programme returns the 95% confidence limit (L MC), were determined with the Monte Carlo simulation, and the P values derived from the parametric were calculated from the concatenated sequences of all five polymorphic markers. We also compared the values of V D and L to assess the population structure of C. cayetanensis using allelic profile data.
Phylogenetic analysis
The sequences of the SSU rRNA gene and the concatenated multilocus sequences were aligned with reference sequences using the programme ClustalW 2.1 (http://www.clustal.org/) to identify the species and genotypes. Maximum likelihood trees were constructed with the programme MEGA 5 (http://www.megasoftware.net/), based on the evolutionary distances calculated with the Kimura two-parameter model. The reliability and robustness of these trees were assessed with a bootstrap analysis of 1000 replicates.
Substructure analysis
A Bayesian cluster analysis was performed on the allelic profile data with the software STRUCTURE version 2.3.4 (http://pritch.bsd.uchicago.edu/software.html) to assess the presence of distinct subpopulations. Twenty simulations were conducted for each value of K (=2–10) using a burn-in period length of 104 and 105 Markov chain Monte Carlo replicates (Pritchard et al. Reference Pritchard, Stephens and Donnelly2000). The most appropriate value for K was calculated using a statistics-based approach implemented in the software Structure Harvester v0.6.94 (Earl and von Holdt, Reference Earl and von Holdt2012).
Statistical analysis and nucleotide sequence accession numbers
The statistical analysis was performed with SPSS software 22.0. The infection rates were compared with a χ 2 test, and differences were considered significant at P < 0·05.
All the nucleotide sequences have been submitted to GenBank at the National Center for Biotechnology Information under the following accession numbers: KY770755–KY770759 for the SSU rRNA gene; KY770752–KY770754 and KY770764–KY770777 for the microsatellite loci.
RESULTS
Molecular epidemiology of C. cayetanensis
The overall incidence of C. cayetanensis infection was 1·2% (76/6579), with 1·6% (50/3173) in Zhengzhou and 0·8% (26/3406) in Kaifeng (P < 0·05), identified with a molecular analysis based on the SSU rRNA gene (Table 1). The male and female infection rates were 1·2% (44/3629) and 1·1% (32/2950) (P > 0·05), respectively. Cyclospora cayetanensis was detected at all age groups, and the infection rate varied from 0·5 to 1·6% (P > 0·05) (Table 1). Longitudinally, the infection rates varied from 0·9 to 1·8% in the years from 2011 to 2015 (Table 2).
Table 1. Distribution of Cyclospora cayetanensis infection in patients in Henan Province, China

Table 2. Longitudinal yearly distribution of the prevalence of Cyclospora cayetanensis infections in 2011–2015

Sequence polymorphisms and multilocus genotypes
Of the 76 specimens positive for the C. cayetanensis SSU rRNA gene, the CYC3, CYC13, CYC15, CYC21 and CYC22 loci were amplified in 49, 59, 57, 52 and 56 specimens, respectively (Supplementary Table 1S). The genetic variation at the five loci involved the lengths of the tetranucleotide TGTA and TATA repeats in CYC3, the trinucleotide GAT repeats in CYC13, the trinucleotide TGC repeats in CYC15, the dinucleotide AT repeats in CYC21 and the dinucleotide AC repeats in CYC22, and in the SNPs outside the tandem repeat regions.
The number of genotypes and gene diversity (H d) at the individual loci were calculated with DnaSP. Generally, marker CYC21 afforded higher resolution than the other markers (Table 3). The intragenic LD and recombination among segregating sites at each locus were assessed with a linear regression analysis. The numbers of pairwise comparisons and significant pairwise comparisons detected with Fisher's exact test and the Bonferroni correction were also calculated. The markers CYC3, CYC15 and CYC22 had complete LD (LD = 1) when the C. cayetanensis isolates from China and other countries were analysed (Table 4).
Table 3. Number of Cyclospora cayetanensis genotypes and nucleotide polymorphisms at five microsatellite loci

a Isolates from China in the present study.
b Isolates from China in another study.
c Isolates from the USA, Nepal, Peru and Spain.
Table 4. Intragenic linkage disequilibrium and recombination events at individual genetic loci

S, number of segregating sites; P, number of pairwise comparisons; F, number of significant pairwise comparisons detected with Fisher's exact test; B, number of significant comparisons after the Bonferroni correction; LD (|D′|), linkage disequilibrium between sites, where X is the nucleotide distance (measured in kilobases, kb); Rm, minimum number of recombination events.
a Among the 61 Cyclospora cayetanensis isolates from China.
b Among the 79 C. cayetanensis isolates from China and other countries.
Multilocus sequence analysis
Forty-five specimens were successfully amplified at all five loci and were therefore used in the population genetic analysis of the MLST data. The multilocus analysis was performed by concatenating the sequences of all five polymorphic markers into a contig of 2607 bp. Together, these 45 isolates formed a total of 29 MLGs (from MLG26 to MLG54) (Supplementary Table 1S).
The interlocus LD calculated with DnaSP was tested over all the segregating sites in the multilocus contigs of all 45 specimens and the 34 reference sequences (Guo et al. Reference Guo, Roellig, Li, Tang, Frace, Ortega, Arrowood, Feng, Qvarnstrom, Wang, Moss, Zhang and Xiao2016). Together, these 79 isolates contained 54 MLGs (MLG1 to MLG54), 64 polymorphic sites, 29 haplotypes, gene diversity H d = 0·920, nucleotide diversity P i = 0·00552 and an average number of nucleotide differences K = 14·382 (Table 5). Of the 1953 pairwise comparisons, 1043 were significant on Fisher's exact test, and 578 of these remained significant after the Bonferroni correction.
Table 5. Genetic diversity in Cyclospora cayetanensis based on the analysis of the concatenated sequences at five genetic loci

N, number of isolates; S, number of polymorphic sites; H, number of haplotypes; H d, haplotype diversity; P i, nucleotide diversity; K, average number of nucleotide differences; F s, Fu's statistic, testing selective neutrality based on genotype frequency; Zns, interlocus genetic association; LD (|D′|), linkage disequilibrium per site; R m, minimum number of recombination events.
a Isolates from China in the present study.
b Isolates from China in another study.
c Isolates from the USA, Nepal, Peru and Spain.
Phylogenetic analysis
A Bayesian statistical approach was used to assess the phylogeny of the 76 positive specimens detected, based on the SSU rRNA gene. All the isolates clustered into the C. cayetanensis clade (Fig. 1). An analysis of the genetic sequence polymorphisms showed that there were minimal (1–2) nucleotide substitutions among the isolates from Mexico (KC662279), Poland (KP642665), China (FJ009129), Nepal (KC662285) and Peru (KC662292) (Supplementary Fig. 1S).

Fig. 1. Phylogenetic relationships of the Cyclospora cayetanensis identified in this study and reference isolates based on SSU rRNA gene sequences. SSU rRNA, small subunit ribosomal RNA.
Three main clusters emerged in a phylogenetic analysis of the 54 individual MLGs based on the concatenated multilocus sequences (Fig. 2). The black clade contains sequences from the USA, Nepal and Peru. However, the isolates from China contributed predominantly to two main clades (red and blue clades), together with one isolate from Spain (2011) and one from Peru (2006).

Fig. 2. Phylogenetic relationships of the 54 distinct MLGs of Cyclospora cayetanensis in humans with circle using concatenated five microsatellite loci sequences. MLG, multilocus genotype.
Population substructure
A population structural analysis was performed based on the allelic profile data from the five loci, using STRUCTURE. The peak value for ΔK occurred at K = 3 (Supplementary Fig. 2S), after a burn-in length of 104 and 105 replicates in 20 Markov chain Monte Carlo simulations. Therefore, 79 isolates (including the 45 specimens detected in this study and 34 reference sequences) of C. cayetanensis formed three subpopulations (K = 3), which are shown with sort and in multiple lines (Fig. 3).
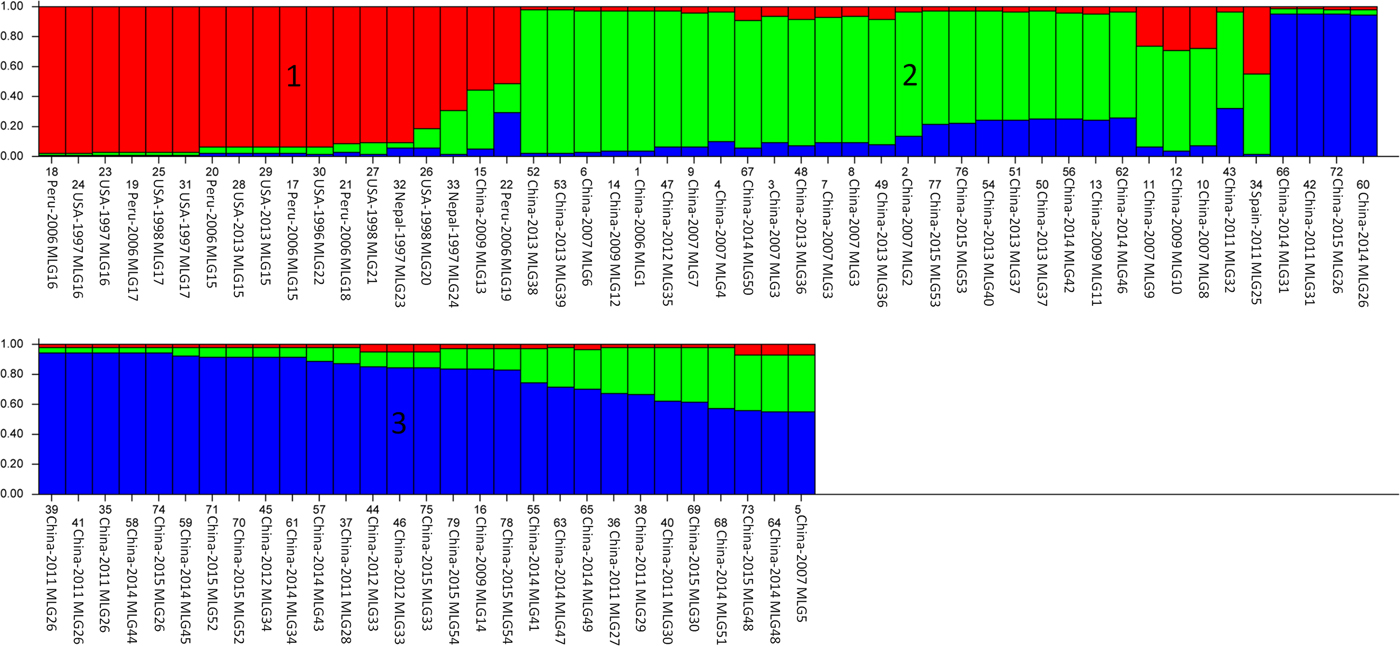
Fig. 3. Population structure inferred with Bayesian clustering using multilocus subtype information for all 79 Cyclospora cayetanensis isolates (K = 3). Lower labels are the isolate numbers, locations, years and multilocus genotypes.
The overall population and individual populations of C. cayetanensis in humans all displayed significant but incomplete LD (Table 5). Although significant LD (|D′| Y = 1·0045 − 0·1025X) was detected, in a further LD analysis per site, the negative slope in the |D′| graph demonstrated a decline in LD with increasing nucleotide distance. This incomplete LD is probably a sign of recombination. To clarify this, a recombination analysis was performed, which yielded a minimum of five recombination events and an estimate 0·001 per gene.
LD was further assessed by calculating the standardized index of association (I A S) based on the allelic profile data for the loci, using the lian software. A zero or negative value of I A S indicates a randomly mating population and that the alleles in the population are in linkage equilibrium (LE), whereas values >0 indicate a non-panmictic population structure, and that the alleles in the population are in LD. A V D < L suggests a panmictic population in LE, whereas V D > L indicates a clonal population in LD (Haubold and Hudson, Reference Haubold and Hudson2000). In the analysis of the allelic profile data from the total 79 isolates, the value of I A S was positive (0·0511) and V D (1·1712) > L (1·0660) (Table 6), indicating that the population was in LD. To test the significance of LD, a Monte Carlo simulation and parametric method were used. They produced a significant P MC value (<0·001) (Table 6), indicating that the population was clonal, with LD.
Table 6. Linkage disequilibrium analysis based on allelic profile data from five genetic loci

N, number of isolates; H, mean genetic diversity; I A S, standardized index of association, calculated with the programme LIAN 3·5; P MC, significance of obtaining this value in 1000 simulations using the Monte Carlo method; V D, variance of pairwise differences; L, 95% critical value of V D; V D > L indicates linkage disequilibrium.
a Isolates from China in the present study.
b Isolates from China in another study.
c Isolates from the USA, Nepal, Peru and Spain.
DISCUSSION
This study demonstrates a frequency of C. cayetanensis of 1·2% (76/6579) in Chinese patients in 2011–2015, and that all age groups were infected (P > 0·05). A similar incidence of C. cayetanensis infection (1·3%, 125/9984) was reported in a hospital in the Honduras (in 2002–2011), based on Ziehl–Neelsen carbol fuchsin staining (Kaminsky et al. Reference Kaminsky, Lagos, Raudales Santos and Urrutia2016). However, a lower infection rate (0·67%, 60/8877) was detected in a paediatric hospital (patients aged <15 years) in Mexico (in 2000–2009), based on morphometric characteristics and PCR (Orozco-Mosqueda et al. Reference Orozco-Mosqueda, Martínez-Loya and Ortega2014). A higher prevalence (3·94%, 20/507) was detected in school children (aged 3–14 years) in Nepal, with modified acid-fast staining (Bhandari et al. Reference Bhandari, Tandukar, Parajuli, Thapa, Chaudhary, Shrestha, Shah, Sherchan and Sherchand2015), and a markedly higher rate was reported in humans in Italy (27·5%, 11/40), based on quantitative PCR (Giangaspero et al. Reference Giangaspero, Marangi, Koehler, Papini, Normanno, Lacasella, Lonigro and Gasser2015).
The occurrence of cyclosporiasis is markedly seasonal throughout the world (Ozdamar et al. Reference Ozdamar, Hakko and Turkoglu2010; Orozco-Mosqueda et al. Reference Orozco-Mosqueda, Martínez-Loya and Ortega2014; Kaminsky et al. Reference Kaminsky, Lagos, Raudales Santos and Urrutia2016), including in China (in July to September) (Zhou et al. Reference Zhou, Lv, Wang, Wang, Jian, Zhang, Ning, Fu, Wang, Qi, Yao, Zhao, Zhang, Sun, Shi, Arrowood and Xiao2011), when it is warm with sufficient precipitation. This intriguing fact is consistent with a report from the Honduras (Kaminsky et al. Reference Kaminsky, Lagos, Raudales Santos and Urrutia2016), whereas the disease reportedly also occurs in warm and dry months in Turkey (Ozdamar et al. Reference Ozdamar, Hakko and Turkoglu2010). Generally, the sporogony of C. cayetanensis oocysts occurs at the appropriate temperature and humidity (Ortega and Sanchez, Reference Ortega and Sanchez2010). The lack of a consistent pattern in the incidence of C. cayetanensis might be attributable to the specific sporulation characteristics of the oocysts in different climates and geographic regions.
The lack of a laboratory culture method for Cyclospora hinders the easy isolation of oocysts, limiting our understanding of its basic biology (Eberhard et al. Reference Eberhard, Ortega, Hanes, Nace, Do, Robl, Won, Gavidia, Sass, Mansfield, Gozalo, Griffiths, Gilman, Sterling and Arrowood2000). However, the analysis of population structures can clarify the epidemiology and evolution of C. cayetanensis (Tibayrenc and Ayala, Reference Tibayrenc and Ayala2014). This MLST analysis of C. cayetanensis provides substantial new insight into the population genetics of C. cayetanensis in humans. Three clusters were identified with a phylogenetic analysis (Fig. 2), and similarly, three subpopulations were detected in a population structural analysis (Fig. 3). Except for two isolates from Spain and Peru, the C. cayetanensis isolates in China were significantly different from those in other countries in our evolutionary analysis. The C. cayetanensis isolates from China formed two main clusters or subpopulations, which may have novel genotypes within the C. cayetanensis.
MLST analysis, which included several mini- and microsatellite markers, identifies panmictic, epidemic and clonal population structures (MacLeod et al. Reference MacLeod, Tweedie, Welburn, Maudlin, Turner and Tait2000). The presence of strong LD and very limited recombination supports the existence of a significant clonal structure in C. cayetanensis in humans (Table 6), whereas Cryptosporidium hominis and T. gondii have typical clonal population structures (Gatei et al. Reference Gatei, Das, Dutta, Sen, Cama, Lal and Xiao2007; Khan et al. Reference Khan, Fux, Su, Dubey, Darde, Ajioka, Rosenthal and Sibley2007) and the Chinese Cryptosporidium andersoni populations have an epidemic structure (Wang et al. Reference Wang, Jian, Zhang, Ning, Liu, Zhao, Feng, Qi, Wang, Lv, Zhao and Xiao2012). Plasmodium falciparum displays different genetic structures at different geographic distributions (Tibayrenc and Ayala, Reference Tibayrenc and Ayala2002; Su et al. Reference Su, Hayton and Wellems2007; Buckee and Gupta, Reference Buckee and Gupta2010). Cryptosporidium parvum displays all these structures, depending on its host and geographic distribution (Mallon et al. Reference Mallon, MacLeod, Wastling, Smith, Reilly and Tait2003; Tanriverdi et al. Reference Tanriverdi, Markovics, Arslan, Itik, Shkap and Widmer2006; Morrison et al. Reference Morrison, Mallon, Smith, MacLeod, Xiao and Tait2008; Wang et al. Reference Wang, Zhang, Axén, Bjorkman, Jian, Amer, Liu, Feng, Li, Lv, Zhao, Qi, Dong, Wang, Sun, Ning and Xiao2014).
In conclusion, we have demonstrated the frequency of C. cayetanensis in patients in Henan Province, China. A clonal population structure with LD and three distinct subpopulations were identified in the human C. cayetanensis isolates. Numerous further surveys and more MLST data from humans and other sources (soil, vegetation and water) in epidemic areas are required for in-depth analyses to identify new genotypes within the C. cayetanensis, and to investigate the population differentiation.
FINANCIAL SUPPORT
This study was supported, in part, by the National Natural Science Foundation of China (U1404327), the Key Program of the National Natural Science Foundation of China (31330079) and the Key National Science and Technology Specific Projects (2012ZX10004220-001).
SUPPLEMENTARY MATERIAL
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182017001299.











