INTRODUCTION
Tuna (Scombridae) are teleost fish with unique biological traits that include thunniform swimming, elevated metabolic rate, and endothermy. They have an extensive capacity for migratory movements and a wide geographical distribution. The largest of the Thunnus species, Bluefin tunas, comprise 3 species (T. thunnus, T. maccoyii, and T. orientalis), which are also the longest lived, and have the widest geographical distributions (Block and Stevens, Reference Block and Stevens2001). Whereas species like yellowfin (T. albacares) prefer tropical waters and either short dives below the thermocline or rapid deep dives to greater depths (Block et al. Reference Block, Keen, Castillo, Dewar, Freund, Marcinek, Brill and Farwell1997), Atlantic (T. thynnus) and Pacific bluefin (T. orientalis) tunas have an increased thermal tolerance (~1·8–30°C) (Block and Stevens, Reference Block and Stevens2001; Marcinek et al. Reference Marcinek, Blackwell, Dewar, Freund, Farwell, Dau, Seitz and Block2001; Kitagawa et al. Reference Kitagawa, Boustany, Farwell, Williams, Castleton and Block2007), reflecting their ability to expand into high latitude, subpolar waters (Block et al. Reference Block, Teo, Walli, Boustany, Stokesbury, Farwell, Weng, Dewar and Williams2005). Tunas represent the most valuable fisheries and finfish aquaculture product currently recognized, with more than half of the world production concentrated in the Mediterranean Sea, and additional ranching operations present in North America and Australia (Miyake et al. Reference Miyake, De la Serna, Di Natale, Farrugia, Katavić, Miyabe and Tičina2003). During the fattening period in sea-cages, tuna in the Mediterranean are fed fresh catches of local purse seiners (European pilchard, Sardina pilchardus; sprat, Sprattus sprattus; European anchovy, Engraulis encrasicolus), as well as imported frozen herrings, Clupea harengus and anchovy, Engraulis encrasicolus from the Atlantic and North Sea.
Long-distance migrations of tuna and their exposure to a wide range of ambient water temperatures facilitate infections with several parasitic groups. Parasite communities of wild and reared bluefin tuna display remarkable diversity, whilst the highest levels of prevalence and abundance are achieved by Didymozoidae (Monticelli, 1888) (Trematoda, Digenea) (Munday et al. Reference Munday, Sawada, Cribb and Hayward2003; Mladineo and Tudor, Reference Mladineo and Tudor2004). Didymozoidae have a wide distribution, occurring in tropical and subtropical areas, with 23 species occurring only in Mediterranean fish (Nikolaeva, Reference Nikolaeva and Hargis1985). Didymozoids are gonochorists or hermaphrodites, mostly encysted in pairs, with a wide and species-specific distribution in host tissues. In bluefin tuna, they are found to inhabit different niches, parasitizing gills and connective tissue; mucous membranes in the nose, mouth, ocular and gill cavities, digestive organs, kidneys, gonads, skin and fins (Yamaguti, Reference Yamaguti1958, Reference Yamaguti1970). Within different species of didymozoids there is often an enormous difference in body size and localization in the host that reflects the range of histopathological changes induced in the host; from negligible host reaction, to necrosis and sloughing of gill epithelium with secondary bacterial infections (Mladineo, Reference Mladineo2006; Di Maio and Mladineo, Reference Di Maio and Mladineo2008). Little is known about their life cycle, except the existence of many intermediate hosts (small pelagic fish and cephalopods) that propagate the metacercariae to teleosts (Lester and Newman, Reference Lester and Newman1986). Likewise, didymozoid larvae have been isolated encysted in the musculature of a large number of cephalopods and fish species (Yamaguti, Reference Yamaguti1970), some of them occurring in the tuna diet both in the wild and under farming conditions.
Different components distinguished by varying degrees of separation can be recognized within populations of most marine organisms. For example, 2 sympatric components may have a common feeding area but separated spawning areas, while others may be separated to a greater extent (MacKenzie, Reference MacKenzie and Rohde2005). Parasites can be used to study host populations and their migrations. This is especially important in large, fast-swimming, predatory, pelagic teleosts, where appropriate knowledge of their population characteristics is a prerequisite for efficient stock management and decision-making. Holt and Boulinier (Reference Holt, Boulinier, Thomas, Renaud and Guégan2005) assumed that the distribution of hosts in the environment will condition the distribution of their parasites, implying that highly migratory fish hosts with cosmopolitan distribution will reflect the cosmopolitanism of their parasites. However, despite some studies that have proved the existence of cryptic species (e.g. Miura et al. Reference Miura, Torchin, Kuris, Hechinger and Chiba2006) or sympatric speciation (see Huyse et al. Reference Huyse, Poulin and Théron2005; Mattiucci and Nascetti, Reference Mattiucci and Nascetti2008), there are few studies based on molecular approaches that demonstrate the cosmopolitanism of platyhelminths isolated from long-distance migrating teleost species. General trends in parasitology tend to put more emphasis on the discovery of cryptic speciation with intriguing evolutionary pathways, rather than on parasites that have overcome ecological barriers and have succeeded in inhabiting transoceanic areas without undergoing speciation. Given our extensive knowledge of parasites in pelagic fish, one would assume that robust data exist on the distribution of these parasites. However, there are only a few examples of such studies. For the 3 platyhelminths species (Cardicola forsteri, Hexostoma thynni and Capsala sp.) considered pathogenic for farmed Southern Bluefin tuna, Aiken et al. (Reference Aiken, Bott, Mladineo, Montero, Nowak and Hayward2007) found remarkable evidence based on 28S and ITS-2 nuclear ribosomal (rDNA) sequences for their worldwide distribution shared by Pacific, Atlantic, Southern bluefin (T. macoyii) and yellowfin tuna (T. albacares). Similarly, cosmopolitanism was shown for plerocercoids of Tentacularia coryphaenae (Cestoda: Trypanorchyncha), which had less than 1% of mitochondrial DNA (mt) sequence variation between parasites from southern Java coastal stocks and North Atlantic sampled from different host species (Palm et al. Reference Palm, Waeschenbach and Littlewood2007). Interestingly, reviewing extensive literature on groupers' (Serranidae) trematodes, Cribb et al. (Reference Cribb, Bray, Wright and Pichelin2002) listed only 2 species with a cosmopolitan ‘affinity’. The authors, however, advised caution, since molecular tools were not used in these studies to discern possible cryptic species. This remarkable ‘breadth’ of some parasites needs to be more carefully examined, since the ecological constraints that they encounter during their life cycle are especially strong, constantly demanding the balancing of parasite fitness.
The aim of this study was to evaluate phylogenetic structure of Didymozoidae members shared by Pacific and Atlantic bluefin tunas, inferred from fragments of 3 loci with different evolutionary speed rate. We used 2 ribosomal DNA loci (28S and ITS-2) that have already proven to be reliable tools for interspecific characterization of parasites (Anderson and Barker, Reference Anderson and Barker1993; Kaci-Chaouch et al. Reference Kaci-Chaouch, Verneau and Desdevises2008), and a mitochondrial gene (cytochrome oxidase 1, cox1) whose rapid evolutionary rate enables differentiation of parasite populations at the intraspecific level (Valentini et al. Reference Valentini, Mattiucci, Bondanelli, Webb, Mignucci-Giannone, Colom-Llavina and Nascetti2006; Mattiucci et al. Reference Mattiucci, Paoletti, Damiano and Nascetti2007). This approach will increase our knowledge of didymozoid zoogeography as well as identify those didymozoid species that could be successfully employed as biological tags for the stock assessment studies of their hosts.
MATERIALS AND METHODS
Fish and tissue sampling
Atlantic bluefin tuna (Thunnus thynnus) were caught from the wild in May 2005/2006, in waters off the Island of Jabuka (43°05″67′N, 15°28″63′E) in the Adriatic Sea. They were then trawled to the farm and fed with mixed fish from small trawling boats, combined with frozen imported herring. Gills and visceral organs were collected in 2005 and 2006 from daily juvenile tuna mortalities and harvested fish (Wt=29·21±15 kg in 2005 and Wt=25·18±14 kg in 2006) at the facility on the NW part of the Island of Brač (Croatia) in the Adriatic Sea.
Pacific bluefin tuna (T. orientalis) were captured by hook and line from the Mexican farming cages off San Diego, CA, USA, in waters of NW Mexico, held on board the fishing vessel in wells filled with flowing seawater, and transported by truck to the Tuna Research and Conservation Center (TRCC) in Pacific Grove, CA, USA (36°37′6″N, 121°54′10″W). Fish were held in two 109 m3 circular tanks containing seawater. Fish were euthanased by pithing and necropsy was done immediately after death in order to isolate and identify didymozoids.
Parasitological examination was performed as previously described (Mladineo et al. Reference Mladineo, Žilić and Čanković2008). Briefly, didymozoid cysts from both tuna hosts were collected from gill and visceral organs, measured and ruptured with fine needles under the stereomicroscope. Individuals were flattened under cover-slip pressure, stained in Borax carmine and mounted in Canada balsam. Identification was performed following Ishii (Reference Ishii1935), Yamaguti (Reference Yamaguti1958, Reference Yamaguti1970) and Pozdnyakov and Gibson (Reference Pozdnyakov, Gibson, Bray, Gibson and Jones2008). For phylogenetic analysis, specimens were fixed in absolute alcohol and stored at 4°C.
DNA isolation and polymerase chain reaction (PCR)
For phylogenetic analysis, genomic DNA was isolated using the QIAGEN DNeasy Blood and Tissue Kit (Qiagen). The 3 loci of interest were amplified using 0·8 μm of each of the following primers: for 28S rDNA (~673 bp) forward primer 5′ GTCCGATAGCGAACAAGTACCGT 3′ and reverse primer 5′ AGCATAGTTCACCATCTTTCGGGTCTCAA 3′; for the partial ITS-2 (~1·5 kb) forward 5′ GTCGTAACAAGGTAGCTGTA 3′ and reverse primer 5′ TATGCTTAAGTTCAGCGGGT 3′; and for the mitochondrial cox1 (~800 bp) was amplified by forward primer 5′ TTTTTTGGGCATCCTGAGGTTTAT 3′ and reverse primer 5′ CAACAAATCATGATGCAAAAGG 3′. The rest of the reaction mix consisted of 1·75–2·2 mM of MgSO4, 220 μm of dNTP, 22 U/ml of PCR SuperMix High Fidelity DNA polymerase mixture (Invitrogen) and 3 ng/μl of template. The amplification profile for 28S rDNA consisted of initial denaturation for 30 s at 94°C, 35 cycles of denaturation for 30 s each at 94°C, annealing at 58°C for 30 s, elongation for 60 s at 72°C with final extension of 10 min at 72°C. For ITS 2 and cox1: initial denaturation for 30 s at 94°C, 35 cycles of denaturation for 30 s each at 94°C, annealing at 56°C for 90 s, elongation for 90 s at 72°C with final extension of 10 min at 72°C.
DNA sequencing and alignment
PCR products were purified using QIAquick PCR Purification Kit (Qiagen) and sequenced on an ABI 3100 automatic DNA sequencer (Applied Biosystems), using the ABI PRISM BigDye Terminator Cycle Sequencing Kit, in both directions. Sequences were aligned with other Didymozoidae or Hemiuridae sequences stored in GenBank (http://www.ncbi.nlm.nih.gov/Genbank/GenbankSearch.html), by Clustal X, implemented in the MEGA 3.1 software, using default parameters. Bayesian Inference (BI) analysis (Larget and Simon, Reference Larget and Simon1999) was performed on the full consensus sequences using MrBayes v3.1.2. (Huelsenbeck and Ronquist, Reference Huelsenbeck and Ronquist2001). General Time Reversible model with proportion of invariable sites and a gamma-shaped distribution of rates across sites was chosen as a best fit model after analysis in Modeltest (http://darwin.uvigo.es/software/modeltest.html) (nrates=invigamma), having 6 substitution types (nset=6). Four incrementally heated Markov Chains were run for 2 000 000 generations (ngen=2 000 000), sampling every 100 generations (samplefreq=100), where 5000 samples were discarded (burnin=5000). Default values were used for MCMC parameters. Bootstrap values exceeding 0·7 were considered well supported (Hills and Bull, Reference Hills and Bull1993). The same parameters were used to analyse a partitioned data set comprising 3 target genes. Since in a few taxa, sequences of all 3 loci were not obtained, their sequences were assigned as missing data.
A 50% majority rule consensus tree was constructed from the tree output files produced in the Bayesian Inference analysis using TreeView (http://taxonomy.zoology.gla.ac.uk/rod/treeview.html). Sequences were added to GenBank and the Accession numbers of sequences obtained in this study are shown in Table 1.
Table 1. Designated GenBank numbers for didymozoid fragment of 28S rDNA (673-bp), ITS-2 (143-bp) and cox1 (745-bp) locus sequenced in this study
(N/A – not analysed. Assigned letter A stands for specimens isolated from Adriatic T. thynnus, and M for specimens isolated from Mexican T. orientalis.)

RESULTS
Characterization of nuclear and mitochondrial DNA sequences
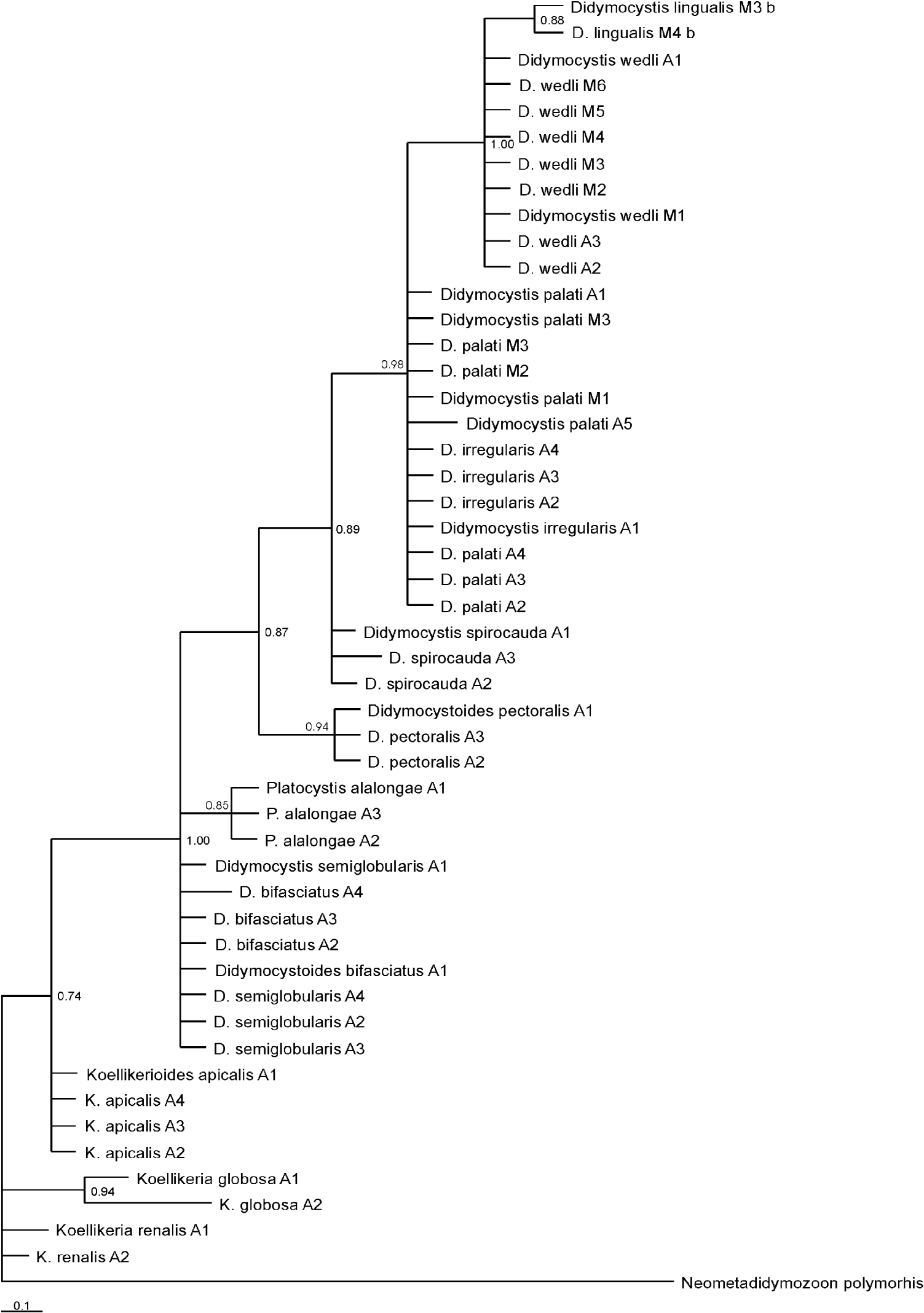
A 673 bp fragment of the 28S locus was sequenced for 65 specimens of Didymozoidae, belonging to 13 species. Didymozoon scombri (AY222195.1) from Scomber scombrus; Didymozoid sp. 1 (AY222194.1) from Apogon cookii, Didymozoid sp. 2 (AY222193) from Epinephelus cyanopodus and Didymozoid sp. 3 (AY222192) from Taeniura lymma, were used for rooting the tree. Genetic divergence based on p-distance values was evaluated. The highest level of interspecific genetic distance was found between Didymocystis wedli and Koellikerioides apicalis (0·103), while the mean overall distance was 0·034. On average, sequences consisted of T 26·1%, C 22·5%, A 21·0% and G 30·4%. The frequency of identical base pairs through all taxa was 613, whereas frequency of transitional pairs was 16 and 6 for transversional pairs. The ratio of transition/transversion was 2·6. Substitution patterns were homogenous among lineages, except between Koellikeria globosa and Didymocystoides pectoralis (ranging from 0·035 to 0·046) and K. globosa and Platocystis alalongae (ranging from 0·027 to 0·049), respectively. The higher difference in base composition biases than expected, based on evolutionary divergence between sequences and by chance alone, was found between K. globosa and K. renalis (0·118), while the average composition distance was 0·028. Maximum composite likelihood (MCL) estimate of the pattern nucleotide substitution showed the probability of substitution for G-A to be 12·46; C-T 23·19; T-C 20·07; and A-G 17·97. The consensus tree constructed based on BI analysis is shown in Fig. 1.
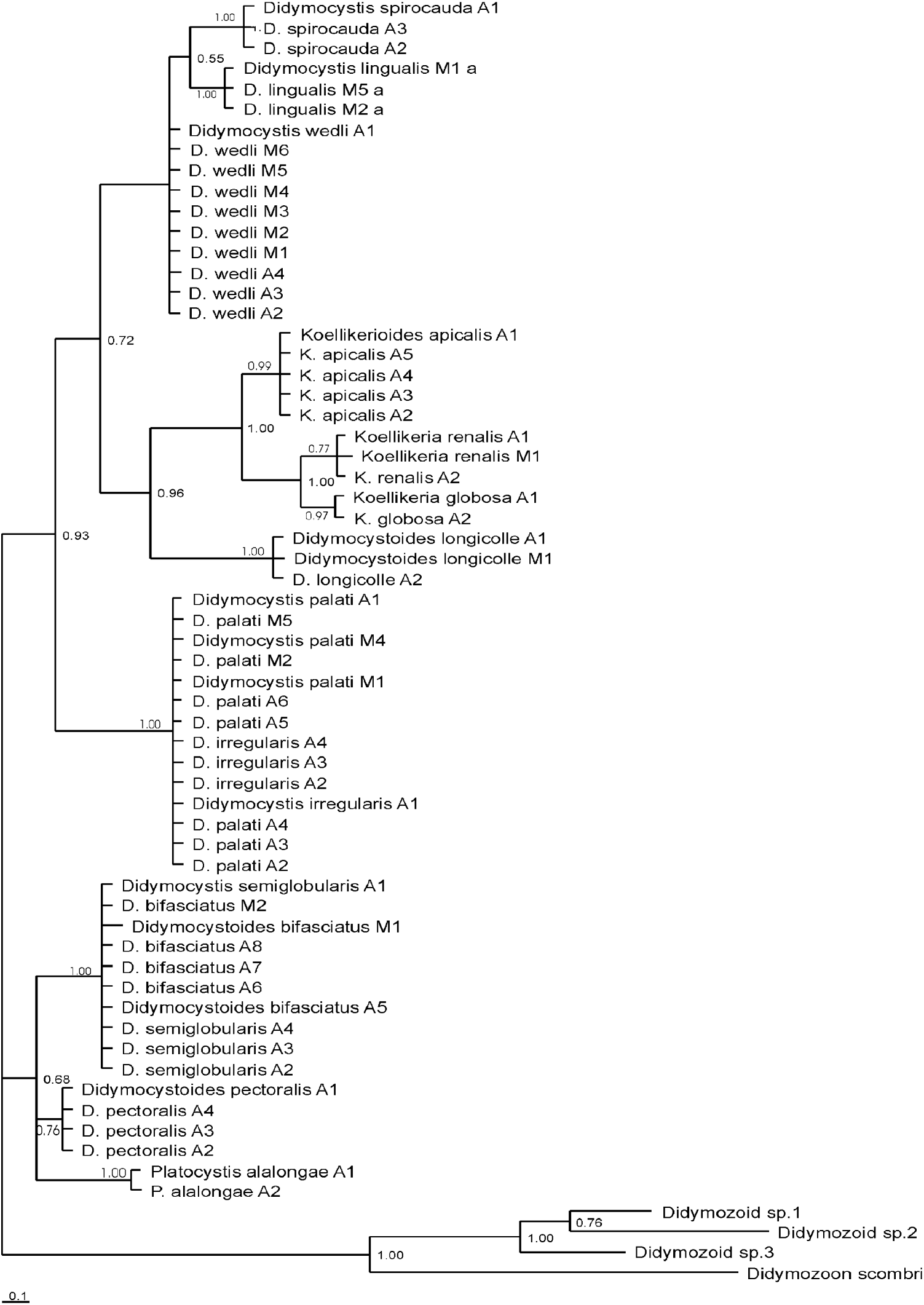
Fig. 1. Phylogenetic tree for didymozoid-based Bayesian Inference (BI) analysis of 28S rDNA sequence data. M at the end of species name stands for Mexican waters, and A for Adriatic, roman number is for the sample number.
A 143-bp fragment of ITS-2 was sequenced for 49 specimens of Didymozoidae, belonging to 12 species. Neometadidymozoon polymorphis (AJ224760) was used for rooting the tree. Genetic divergence based on p-distance values was evaluated. The highest level of interspecific genetic distance was found between K. globosa and D. spirocauda (0·087–0·086)/D. pectoralis (0·074), while the average through all taxa was 0·0290. On average, sequences consisted of T 36%, C 20·8%, A 16·8% and G 26·4%. The frequency of identical base pairs through all taxa was 158, whereas frequency of transitional pairs was 3 and 1 for transversional pairs. Substitution patterns were homogenous among lineages, except between K. globosa and D. palati (ranging from 0·044 to 0·046)/D. irregularis (0·039–0·049)/D. wedli (0·044); between P. alalongae and D. spirocauda (0·008–0·015)/ D. palati (0·010–0·017)/ D. irregularis (0·008–0·017); and between D. bifasciatus and D. spirocauda (0·013–0·019). The higher difference in base composition biases than expected based on evolutionary divergence between sequences and by chance alone, was found between K. globosa and D. spirocauda (0·092)/ D. palati and D. irregularis (0·104)/ D. wedli (0·117), while the average composition distance was 0·009. Maximum composite likelihood (MCL) estimate of the pattern nucleotide substitution showed the probability of substitution for G-A to be 5·29; C-T 37; T-C 21·45; and A-G 8·31. The consensus trees constructed based on BI analysis is shown in Fig. 2.
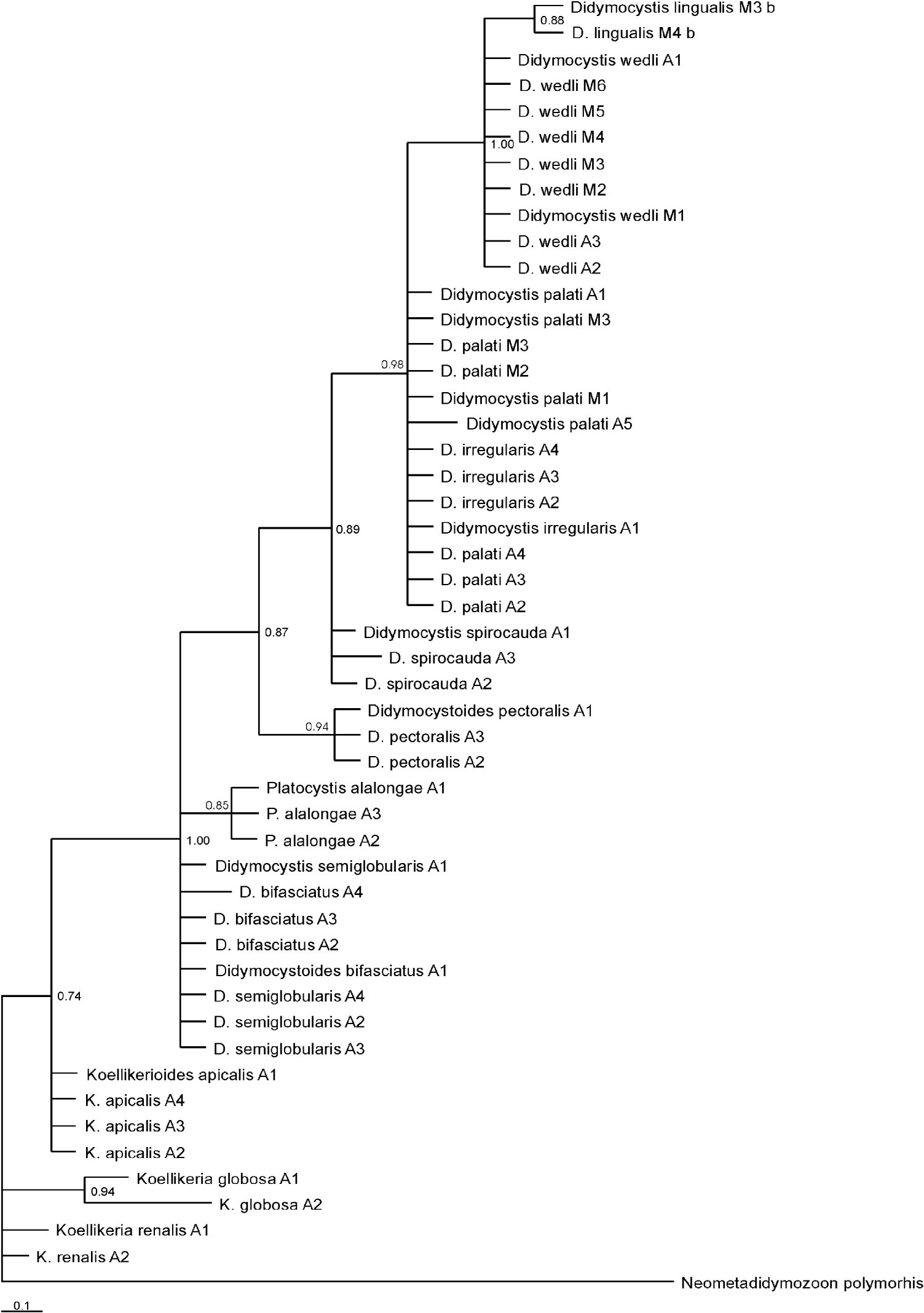
Fig. 2. Phylogenetic tree for didymozoid-based Bayesian Inference (BI) analysis of ITS-2 sequence data. M at the end of species name stands for Mexican waters, and A for Adriatic, roman number is for the sample number.
A 745-bp fragment of cox1 was sequenced for 54 specimens of Didymozoidae, belonging to 13 species. Since no cox1 sequence from taxa closely related to Didymozoidae family was found in GenBank, subfamily Koellikeriinae was used for rooting the tree. Genetic divergence based on p-distance values was evaluated. The highest level of interspecific genetic distance was found between D. pectoralis and D. spirocauda (0·087–0·086); K. renalis and P. alalongae (0·329); and D. wedli and P. alalongae (0·324–0·329), while the average through all taxa was 0·223. On average, sequences consisted of T 41·1%, C 15·7%, A 22% and G 21·1%. The frequency of identical base pairs through all taxa was 531, whereas frequency of transitional pairs was 77 and 74 for transversional pairs. Substitution patterns were heterogeneous among the majority of lineages, where probability was shown to be less than 0·05. The higher difference in base composition biases than expected, based on evolutionary divergence between sequences and by chance alone, was found between P. alalongae and D. pectoralis (4·649–4·710); and D. wedli and P. alalongae (4·363–4·419), while the average composition distance was 0·997. Maximum composite likelihood (MCL) estimate of the pattern of nucleotide substitution showed the probability of substitution for G-A to be 10·99; C-T 26·16; T-C 10·01; and A-G 10·53. The consensus tree constructed based on BI is shown in Fig. 3.

Fig. 3. Phylogenetic tree for didymozoid based Bayesian Inference (BI) analysis of cox1 sequence data. Note encircled differences in 2 populations of Mexican D. wedli and Adriatic Sea' D. wedli, as well as Mexican D. palati and Adriatic Sea' D. palati. M at the end of species name stands for Mexican waters, and A for Adriatic, roman number is for the sample number.
Phylogenetic analysis
Bayesian Inference (BI) analysis inferred from partitioned data sets of all 3 genes analysed together, demonstrated that most didymozoids lie within 1 large clade (Fig. 4). The second smaller, branching sister clade harbours members of the Koellikeriinae subfamily with the sac-like hindbody along with didymozoid from tuna gills with a funnel-like shaped hindbody (D. longicolle). Within the former clade, 2 principal subgroups emerge. The larger subgroup with pronounced polytomy is composed of a clade harbouring didymozoids inhabiting soft head tissues: gills (D. wedli), tongue (D. lingualis) and nerve tissue (D. spirocauda) and the second clade incorporating species from the operculum and cartilaginous parts (D. palati and D. irregularis). The other less robust subgroup branches distinctively into a clade composed of fin parasites (D. pectoralis) and the clade of parasites from the body surface (P. alalongae), operculum (D. bifasciatus) and cartilaginous parts (D. semiglobularis). When 3 loci were analysed separately by BI, the resulting trees (Figs 1, 2 and 3) showed mainly congruent topologies to each other, as well as to the tree from the partitioned data set (Fig. 4), with similar support of nodes. Interestingly, clustering of members of the Koellikeriinae subfamily with the funnel-like shaped D. longicolle was supported by each gene. The only difference was in the relative position of D. longicolle and the Koellikeriinae relative to other taxa. With 28S rDNA (Fig. 1) these taxa clustered with other didymozoids, the 2-lobed, heart-shaped species (D. wedli, D. spirocauda, D. lingualis), but not with ITS-2 or cox1.

Fig. 4. Phylogenetic tree inferred by Bayesian Inference (BI) analysis of partitioned data (28S rDNA, ITS-2, cox1). Grey areas indicate didymozoid species that inhabit the same parasitizing site. M at the end of species name stands for Mexican waters, and A for Adriatic, roman number is for the sample number.
DISCUSSION
For most parasitic groups, morphology is the leading factor that reflects the structuring of phylogenetic relationships. In rare examples, however, habitat has been suggested as one of the primary factors affecting parasite evolution (e.g. Brusca, Reference Brusca1981; Vossbrinck and Debrunner-Vossbrinck, Reference Vossbrinck and Debrunner-Vossbrick2005; Jones et al. Reference Jones, Miller, Grutter and Cribb2008; Ketmaier et al. Reference Ketmaier, Joyce, Horton and Mariani2008). Our data suggest that didymozoids belong to the latter category of parasites, in which habitat selection has been the leading force in shaping phylogenetic relationships. During their evolution, didymozoids have spread and inhabited a remarkable number of different sites of infection, colonizing exterior as well as strictly interior niches, while undergoing evolutionary changes to both the site of infection and gross morphology of the hindbody. This has resulted in the clustering of didymozoids with a heart-shaped hindbody occupying interior niches (D. wedli, D. spirocauda, D. lingualis, D. palati and D. irregularis), apart from the globular, spherical, saccular group of didymozoids on distal or exterior surfaces (D. pectoralis, P. alalongae, D. semiglobularis and D. bifasciatus). This species ecological and phylogenetic ‘segregation’ of didymozoids might be triggered by past interspecific competitions. Poulin (Reference Poulin1998) explained that evolutionary niche restriction with subsequent phylogenetic diversification usually results from parasite interspecific competition, when abundant and prevalent species share the same host for numerous generations and their competition leads to reduction of host fitness. In these conditions, parasite species exert selective pressure on one another and co-evolve in a way that their niches diverge, resulting subsequently in phylogenetic divergence. In contrast, species without competitive tendencies remain at the same site, phylogenetically clustering together within sister clades. However, this competition theory can be challenged since Rohde (Reference Rohde1991) emphasized that competition plays little role if parasite communities are far from being saturated.
More intriguing is the fact that 2 pairs of morphologically very distinctive species (D. palati and D. irregularis; and Didymocystoides bifasciatus and D. semiglobularis, respectively) that occupy the same niches (operculum and cartilaginous tissue of gills and mouth) show no differences in the 28S and ITS-2 sequences. Whereas the hindbodies of D. irregularis and D. palati share the same basic 2-lobed heart-shape, the hindbody of the former has a markedly undulating outline, and in the latter it has a smooth and more ellipsoid shape. Yamaguti (Reference Yamaguti1970) differentiated 2 species based on ovary and vitellaria; in D. irregularis there are 10 or more terminal branches of the ovary and 16 terminal branches in vitellaria, while D. palati has only 2 main ovary branches (with 1 having a side branch), and vitellaria with 3–4 undivided main branches, making identification relatively easy. In the case of the other species pair, the hindbody is of basic globular shape. The hindbody of D. semiglobularis, however, is smooth, and slightly triangular, whereas in D. bifasciatus it has a marked concave impression. When Yamaguti (Reference Yamaguti1970) erected Didymocystoides with the type species D. bifasciatus, he suggested transferring D. semiglobularis into this genus based on its morphology. Since the original description of Ishii (Reference Ishii1935) is lacking detailed description of the genital complex, it is hard to draw comparisons between species on features other than hindbody shape. Although the tree inferred from cox1 data enabled a good resolution of fine phylogenetic variations, we were not able to discern interspecific differences in these cases. This may suggest that this species has morphologically changed in order to adapt to its particular habitat, while molecular differences are visible only at the level of fast-evolving genes.
As a separate subfamily from Didymozoiinae, Koellikeriinae has a distant topology in the phylogenetic structure. Its biological characteristics differ greatly from the other Didymozoiinae as it is one of the few gonochorists in the Didymozoidae family, where tiny males are lodged in a hole of the globular or reniform large female hindbody. Koellikerioides from the tips of gill rakers (K. apicalis) branched separately from Koellikeria from oesophagus (K. globosa) and kidney (K. renalis), reflecting differences in the shape of the hindbody as well as the affinity for soft versus thick connective tissue within the genus. Surprisingly, D. longicolle, a funnel-shaped didymozoid from elongated gill cysts, branched separately from other Didymozoiinae taxa and formed a sister clade with Koellikeriinae.
Analysis of phylograms of 3 didymozoid loci inferred by BI separately as well as partitioned in a single data set, showed the same topology over the sampled taxa. Differences in the tree branching (e.g. position of some internal nodes) can be attributed to the differences in the evolutionary rates between the analysed loci. Olson and Tkach (Reference Olson and Tkach2005) reported that because of the faster evolving nature of mitochondrial genes they are generally not as useful as nuclear ribosomal DNA data in studies of platyhelminth systematics, due to the presumed antiquity of the group. Vilas et al. (Reference Vilas, Criscione and Blouin2005) proposed that mitochondrial DNA was more suited to discerning cryptic speciation. However, our analysis has shown that there is a congruence of phylogram topology with 3 different loci, and for this study the use of nuclear ribosomal and mitochondrial cox1 data is informative for the purpose of determining the phylogenetic structure of the Didymozoinae.
In most host-parasite associations studied to date, phylogenetic congruence is either imperfect or absent, and most associations represent a combination of co-speciation and host-switching (Rannala and Michalakis, Reference Rannala, Michalakis and Page2002). However, the life-history features of both host and parasite, including host specificity and mobility, influence these macro-evolutionary trends (Nadler, Reference Nadler1995; Huyse et al. Reference Huyse, Poulin and Théron2005). In a ruminant host-parasite system, for example, mobility of the host can influence gene flow. More genetic differentiation is displayed in parasites of wild ruminants with a restricted movement array, implying isolation by distance, than in domestic ruminants that are moved across the country by human transportation (Blouin et al. Reference Blouin, Yowell, Courtney and Dame1995). In contrast to terrestrial systems, relatively few investigations of parasites in aquatic systems have demonstrated that wider ranging hosts have parasites of a more homogeneous genetic structure (see Aiken et al. Reference Aiken, Bott, Mladineo, Montero, Nowak and Hayward2007; Palm et al. Reference Palm, Waeschenbach and Littlewood2007). Lo et al. (Reference Lo, Morgan, Galzin and Cribb2001), comparing coral reef fish between French Polynesia and the Great Barrier Reef, reported a lack of molecular variation in digeneans that are separated by more than 6000 km and do not migrate once they complete their pelagic life cycle. Chambers and Cribb (Reference Chambers and Cribb2006) showed that ITS-2 and 28S rDNA sequences of Quadrifoliovarium pritchardae (Digenea: Lecithasteridae) were identical for specimens collected from Exmouth, Australia (Indian Ocean), Heron and Lizard Island, Australia (Western Pacific), and Moorea (Eastern Indo-Pacific). In contrast to coral reef fish, tuna have wide geographical ranges as adults and their mobility may account for the sharing of the same parasites. In addition to the 4 platyhelminths sequenced in the study of Aiken et al. (Reference Aiken, Bott, Mladineo, Montero, Nowak and Hayward2007), 3 copepods (Pseudocycnus appendiculatus, Euryphorus brachypterus and Brachiella thynni) are also known to be cosmopolitan on Thunnus spp., though only on the basis of their morphology, (see Cressey and Cressey, Reference Cressey and Cressey1980; Nowak et al. Reference Nowak, Mladineo, Aiken, Bott and Hayward2006). However, there are few exceptions in the cosmopolitan character of didymozoid species parasitizing tuna, as demonstrated in this study. Inferred from cox1 sequences, it is possible to discern 2 didymozoid species that show marked difference on the intraspecific level between Mexico and Adriatic Sea populations. One is D. wedli, the most frequent species encountered in bluefin tuna parasitizing gill filaments (Mladineo et al. Reference Mladineo, Žilić and Čanković2008), and the other is D. palati, inhabiting cartilaginous parts of the mouth arch which, in comparison to the former, shows a less-distinctive difference in nucleotide sequence. Although D. wedli has a high transmission rate with consequent high abundance that should accelerate its gene flow through populations, suggesting less intrapopulational diversity (Blouin et al. Reference Blouin, Yowell, Courtney and Dame1995), we have found evidence for the opposite. Its isolationist character, as well as its eco-biological characteristics (e.g. easy accessibility for sampling, abundant number per fish and long generation time (MacKenzie, Reference MacKenzie2002)), indicate that D. wedli could be used as a biological marker to differentiate between discrete Atlantic stocks that reach the Gulf of Mexico and the Mediterranean bluefin tuna population (Fromentin and Powers, Reference Fromentin and Powers2005). To confirm this, further research is required as Adriatic tuna sampled in this study were mostly juveniles with a migration range limited to the Mediterranean. The world-wide distribution of the other didymozoids may be attributed to the fact that the distribution of T. orientalis and T. thynnus is considered sympatric, as the main spawning ground of Atlantic bluefin is currently assumed to be along the Mediterranean Sea and the Gulf of Mexico (Frometin and Powers, Reference Fromentin and Powers2005), where Pacific bluefin is fished and farmed (Block and Stevens, Reference Block and Stevens2001). Such mixing of Thunnus spp. provides the opportunity for dispersal of their parasitic fauna over a wider geographical area. This is further enhanced by potentially similar or overlapping distributions of intermediate hosts of didymozoids with Thunnus spp. The Didymozoidae is one of the most taxonomically complex digenean families (see Blair et al. Reference Blair, Bray and Barker1998), abundant in synonyms where fine-tuned identification based on a genital complex has been sometimes obscured by different sizes of hindbodies and subjective descriptions. This makes it difficult to reach taxonomic consensus for this group (Nikolaeva, Reference Nikolaeva and Hargis1985). By adding to the didymozoid molecular data existing in public databases, we hope that we have made the first step towards elucidating phylogenetic relationships between members of the Family Didymozoidae.
ACKNOWLEDGEMENTS
This study was done during tenure of a Fulbright Scholarship for the visiting scholar I.M. in 2006/07 and funds from NOAA Grant for Aquaculture Research. The authors are grateful for indispensable technical support of many people involved in tuna farming at the Island of Brac, Croatia, as well to Chuck Farwell, Matt Price, Alexander Norton, Ty Brandt and Luis Rodriguez of TRCC. Invaluable help with editing was provided by Danielle and Thomas Burrow. Appreciation goes also to anonymous referees whose constructive remarks and suggestions greatly improved this manuscript.