INTRODUCTION
Human sleeping sickness in Africa is a puzzling disease. It is caused by the parasite Trypanosoma brucei (see Table 1). Despite the widespread distribution of the tsetse vectors and animal trypanosomiasis, human disease is found in very specific foci and appears as epidemics followed by periods of endemicity (reviewed by Hide, Reference Hide1999). The locations of these foci are very stable over time but occasionally new epidemics emerge in new localities. For example, the Busoga focus of Uganda has had epidemics recorded from 1900 to the present day while a new epidemic has emerged in the nearby Soroti region – an area free of sleeping sickness prior to 1998 (Welburn et al. Reference Welburn, Picozzi, Fèvre, Coleman, Odiit, Carrington and Maudlin2001). An understanding of the factors which cause these epidemics could clearly lead to an understanding of how to eradicate the disease. Sleeping sickness was one of the earliest examples of the application of molecular epidemiology to a parasitic disease and, as will be seen, some important insights have been made into the origins and dynamics of these epidemics. In this review, we will trace the development of molecular epidemiological approaches with a particular emphasis on African sleeping sickness. As the journal Parasitology has played a significant role in publishing these developments it is an appropriate topic for the Centenary Issue.
Table 1. Characteristics of the subspecies of Trypanosoma brucei

MOLECULAR EPIDEMIOLOGY
The molecular epidemiology of parasites describes the application of DNA-based techniques to the understanding of parasite epidemiology and diversity (Hide and Tait, Reference Hide and Tait1991). The approach was prompted by an emerging understanding that parasites could not be easily packaged into groups of organisms that produced defined diseases in defined hosts. Instead, there were complex epidemiological cycles involving multiple hosts, sometimes multiple diseases, reservoir hosts, specific strains, emerging drug resistance, genetic exchange between strains and even the apparent emergence of new parasite species. Thus tools were needed which could be used to gain a higher level of resolution than morphology in defining the organisms involved. In fact, the discipline of molecular epidemiology strictly emerged from several earlier strands of research. Principally this was the use of phenotypic approaches such as isoenzyme (or allozyme) analysis as a tool to study the epidemiology of parasitic infections. One of the key epidemiological questions in parasitology is determining the correct identity (diagnosis) and taxonomic level of a given parasite (e.g. species, subspecies, strains, variants). This has often proved challenging especially in the case of microscopic parasites (e.g. parasitic protozoa) where morphological distinctions are not easily identified. Two linked problems exist – defining the scope of a taxonomic unit and establishing unambiguous markers for each unit. The scope of a taxonomic unit could be relatively easily established in the absence of any form of gene exchange between units such as occurs in clearly distinct species or clonally reproducing parasites. However, in earlier studies the precise identity of parasitic species or the role of gene exchange was often not known. An inability to clearly define taxonomic units therefore makes it difficult to identify markers that can be used diagnostically to distinguish that taxonomic unit. There is unfortunately a degree of circularity in the problem. There are two levels of identification needed (1) diagnosis – generally referring to diagnostic tools capable of discriminating to the species level and defining a particular association between causative agent and disease and (2) epidemiological tools – generally referring to markers that can be used at the subspecies or strain level to investigate detailed interactions within parasite transmission cycles. This problem of circularity in developing appropriate markers can be overcome by using independent markers that are not linked to the, often very subjective, medical criteria by which parasites were traditionally classified. DNA-based molecular methods offered a potential solution to these problems.
FROM BIOCHEMICAL TO MOLECULAR EPIDEMIOLOGY
The advent of protein-based technologies such as gel electrophoretic separation of isoenzymes (allozymes) enabled distinction of morphologically identical organisms and could be used as a tool for genetic studies. These approaches were initially used with the free-living protozoa both as markers for studying genetic exchange and for defining species (previously defined as syngens) as illustrated by work with Paramecium aurelia (Tait, Reference Tait1970a, Reference Taitb). Key centres in Edinburgh, London and subsequently Bristol then applied these approaches to investigating the genetics and taxonomy of the medically important protozoa, Plasmodium and Trypanosoma. For example these approaches were used for identifying different levels of taxonomic units (e.g. species, subspecies and strains) in Plasmodium (Carter and Walliker, Reference Carter and Walliker1975) and Trypanosoma spp (Kilgour and Godfrey, Reference Kilgour and Godfrey1973; Miles et al. Reference Miles, Toye, Oswald and Godfrey1977; Godfrey, Reference Godfrey1978; Gibson et al. Reference Gibson, Mehlitz, Lanham and Godfrey1978, Reference Gibson, Marshall, De and Godfrey1980; Tait et al. Reference Tait, Babiker and Le Ray1984, Reference Tait, Barry, Wink, Sanderson and Crowe1985). Furthermore, using these techniques, genetic exchange was shown to occur in Plasmodium spp. (Walliker et al. Reference Walliker, Carter and Morgan1971, Reference Walliker, Carter and Morgan1973) and Trypanosoma spp. (Tait, Reference Tait1980; Gibson et al. Reference Gibson, Marshall, De and Godfrey1980; Jenni et al. Reference Jenni, Marti, Schweizer, Betschart, Le Page, Wells, Tait, Paindavoine, Pays and Steinert1986; Gibson, Reference Gibson1989).
When used to analyse the African trypanosomes causing human sleeping sickness, these approaches revealed a complex taxonomic situation. The classical taxonomy described 3 subspecies, Trypanosoma brucei brucei, (non-human infective) Trypanosoma brucei rhodesiense (East African, human infective) and Trypanosoma brucei gambiense (West African, human infective). Multilocus enzyme electrophoresis (MLEE) studies revealed that there were clearly subgroups or strains within both T.b. gambiense and T.b. rhodesiense as well as a high degree of variation between isolates of T.b. brucei (Gibson et al. Reference Gibson, Marshall, De and Godfrey1980; Tait et al. Reference Tait, Babiker and Le Ray1984, Reference Tait, Barry, Wink, Sanderson and Crowe1985). Furthermore, these studies showed that different levels of taxonomy appeared to be operating – the genetic relationships showed that T.b. gambiense appeared to behave like a separate subspecies while T.b. rhodesiense appeared to be a host range variant of T.b. brucei.
It was becoming apparent that the question of what defines a species in parasitic protozoa (and other micro-pathogens) was a complex one to address. Traditionally, the species concept is based on the presence or absence of genetic exchange between taxonomic units. In the case of T. brucei sspp., the extent of the occurrence of genetic exchange in natural populations was unclear. Tibayrenc et al. (Reference Tibayrenc, Kjellberg and Ayala1990) conducted a re-analysis of MLEE data from a wide range of pathogens including T. brucei and Plasmodium and proposed a clonal theory, based on a range of measures such as high levels of heterozygosity, linkage disequilibrium and over-representation of particular genotypes. In the case of T.b. rhodesiense there was evidence for clonality but the results with T.b. brucei were less convincing. Further analysis of a large collection of isolates from across Africa (Mathieu-Daudé and Tibayrenc, Reference Mathieu-Daudé and Tibayrenc1994) was interpreted as showing limited genetic exchange, although geographical substructuring could be an explanation given the diversity of the origins of the strains. These conclusions conflicted with published data showing the existence of genetic exchange (e.g. Tait and Turner, Reference Tait and Turner1990). Analysis of a large collection of isolates from the Lambwe Valley focus, in kenya, provided a partial resolution of the debate by showing that the population had an epidemic population structure (Maynard-Smith et al. Reference Maynard Smith, Smith, O'Rourke and Spratt1993) i.e. that is there was underlying genetic exchange masked by the local expansion of a small number of genotypes. Further analysis of populations from Cote d'Ivoire, Zambia and Busoga, Uganda (Stevens and Tibayrenc, Reference Stevens and Tibayrenc1995) showed a diversity of population structures with evidence for clonality in some populations but an epidemic structure in others (Uganda). This diversity was also illustrated by MLEE analysis of sympatric stocks collected during a sleeping sickness epidemic in Tororo, Uganda (Fig. 1). In this study, it was demonstrated that the population structure of the T.b. brucei stocks appeared to conform to a random mating population structure while the population structure of T.b. rhodesiense was recognized as epidemic (Hide et al. Reference Hide, Welburn, Tait and Maudlin1994). The debate over the role of genetic exchange in T. brucei was not fully resolved using MLEE.
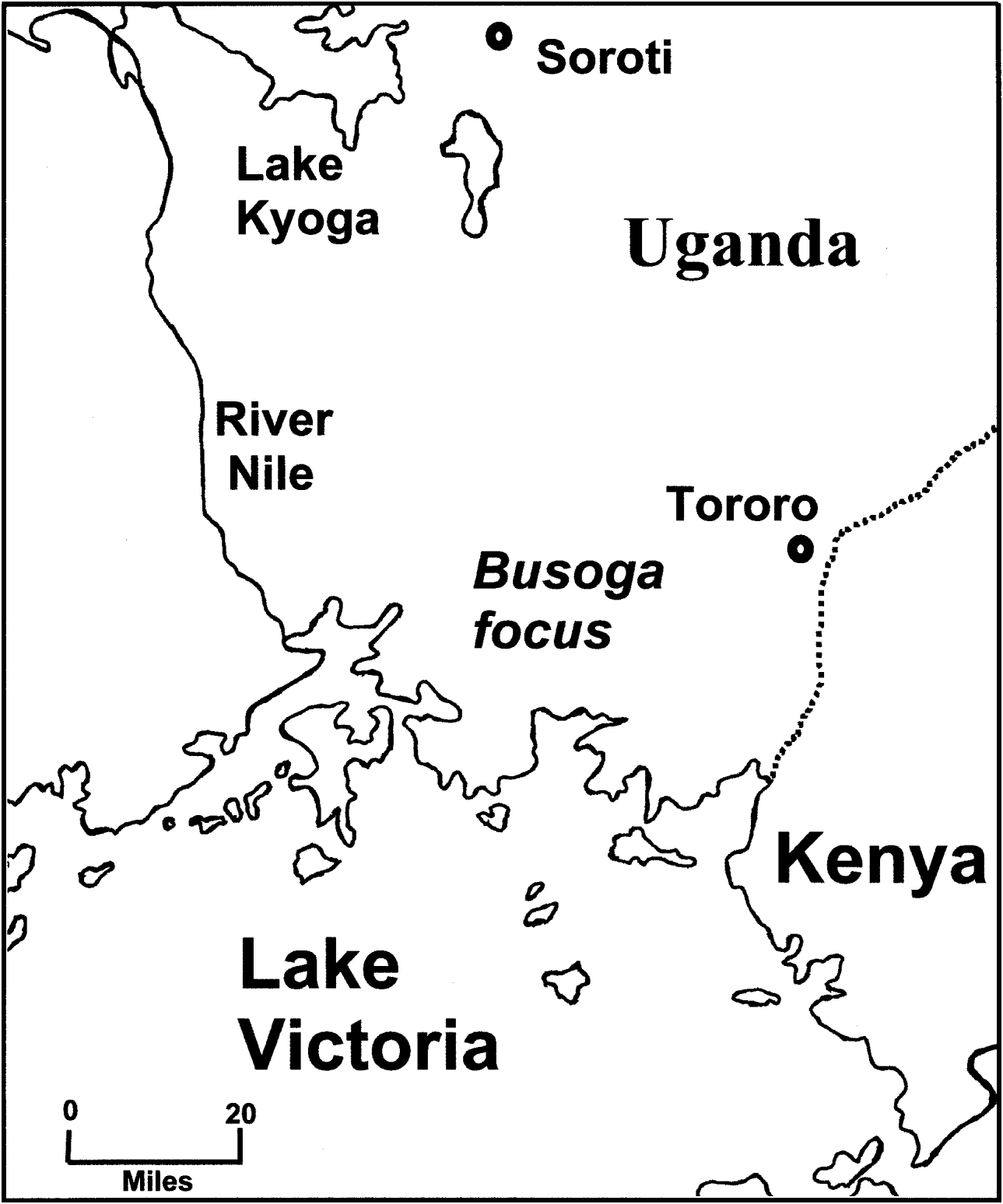
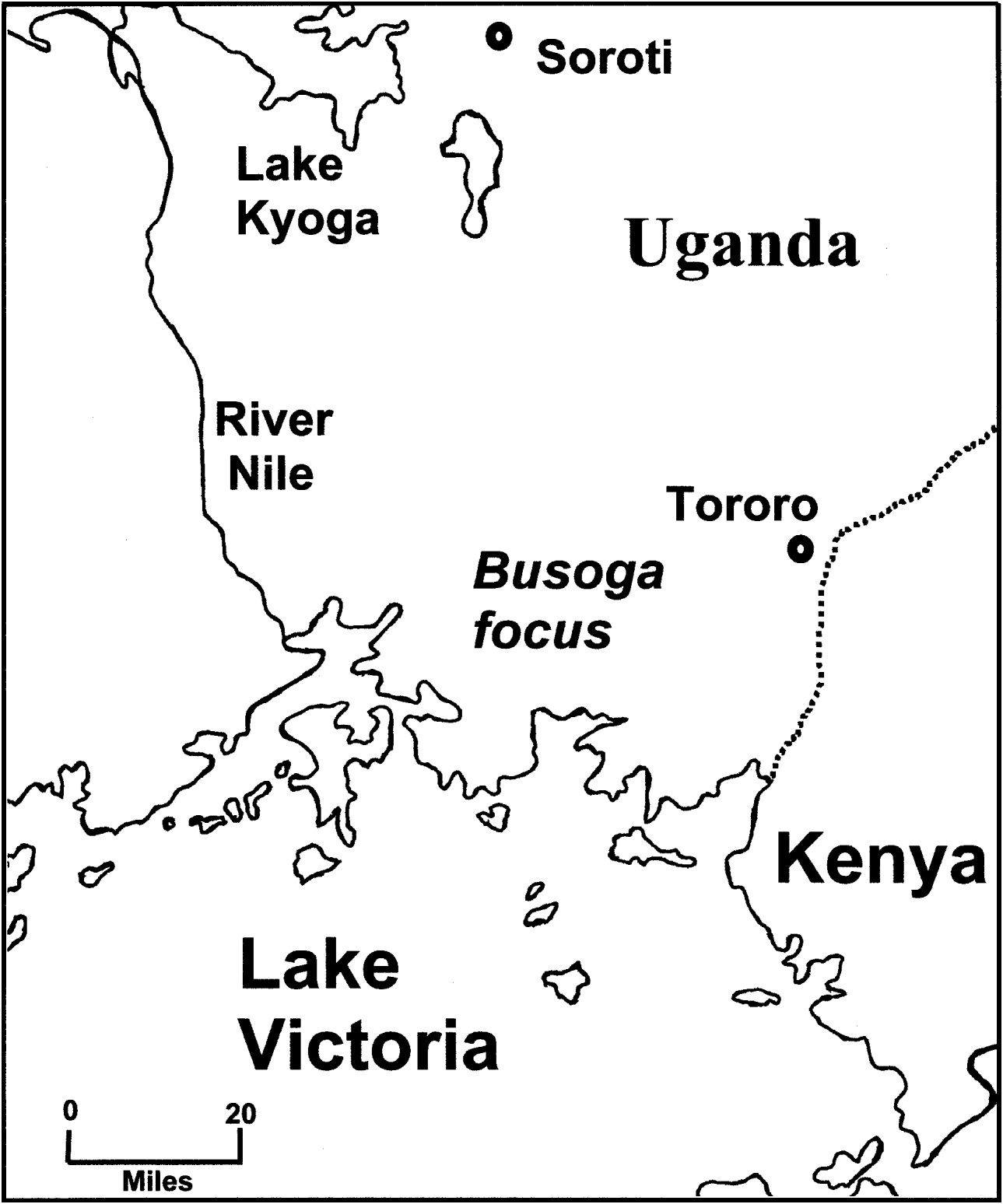
Fig. 1. A map of the Busoga focus of Trypanosoma brucei rhodesiense sleeping sickness in Uganda. Recent epidemics occurred in Tororo (1988 to date) and Soroti (1998 to date).
MLEE is a highly informative technique that enabled epidemiological analyses to be based on a genetic interpretation, with the essential conclusions for African trypanosomes remaining unchallenged today. However, these methods require relatively large quantities of pure parasite material and so are cumbersome for large-scale epidemiological analysis. Initially, blood from patients and animals had to be isolated, followed by several passages through mice (or in culture) and further mouse amplification to produce enough material for analysis. In some cases, the Kit for In Vitro Isolation (KIVI) of trypanosomes has been used to isolate trypanosomes (Truc et al. Reference Truc, Aerts, McNamara, Claes, Allingham, Le Ray and Godfrey1992). During these processes, cloning from individual trypanosomes was also often necessary, with several years work required to analyse an appropriately sized sample set. An additional problem associated with these methods is the potential of the methods to select for particular types of trypanosomes, such as those better adapted to growth in mice, thereby introducing bias into the distribution of parasites recovered (Koffi et al. Reference Koffi, Solano, Barnabé, De Meeûs, Bucheton, Cuny and Jamonneau2007).
Initially, DNA technology was focussed on developing diagnostic tools to identify Trypanosoma species. As it is the subject of another review in this issue (Gibson, Reference Gibson2009), it will not be discussed further here and we will consider the development of DNA based tools for measuring intraspecific variation (Table 2). Initial DNA studies on intraspecific variation in T. brucei were based on analysis of restriction enzyme site variation in the kinetoplast DNA (Borst et al. Reference Borst, Fase-Fowler and Gibson1981). However, with limited stocks available and a relatively small number of restriction enzyme sites covered, it was not possible to discriminate between the 3 subspecies. A larger scale analysis (Gibson et al. Reference Gibson, Borst and Fase-Fowler1985), however, demonstrated that a difference could be detected in stocks originating from East and West Africa. Restriction Fragment Length Polymorphism (RFLP) of VSG antigen genes and numerical taxonomy, based on those RFLPs, revealed that T.b. gambiense was quite distinct from T.b. rhodesiense or T.b. brucei but that the latter two subspecies could not be distinguished (Paindavoine et al. Reference Paindavoine, Pays, Laurent, Geltmeyer, Le Ray, Mehlitz and Steinert1986). However, it was discovered that 2 forms of T.b. gambiense existed (Paindavoine et al. Reference Paindavoine, Pays, Laurent, Geltmeyer, Le Ray, Mehlitz and Steinert1986, Reference Paindavoine, Zampetti-Bosseler, Coquelet, Pays and Steinert1989) one of which was probably the classical T.b. gambiense (Type 1) and one that has been called either non-gambiense or Type 2 or ‘Bouafle’ (Gibson, Reference Gibson1986, Reference Gibson2007; Paindavoine et al. Reference Paindavoine, Zampetti-Bosseler, Coquelet, Pays and Steinert1989). This latter group is relatively rare and may constitute a variant of T.b. brucei that has acquired human infectivity (Gibson, Reference Gibson2007). RFLP methods were developed using repetitive DNA sequences including the ribosomal RNA genes to investigate RFLP variation using multiple loci (Hide et al. Reference Hide, Cattand, Le Ray, Barry and Tait1990). Although these data could not be interpreted genetically and could not detect mixtures of trypanosomes, they offered a highly discriminatory approach to ‘fingerprinting’ trypanosome isolates. The RFLP data combined with numerical taxonomy clearly distinguished the two types of T.b. gambiense and showed that Type 2 appeared to be a subset of West African T.b. brucei strains (Hide et al. Reference Hide, Cattand, Le Ray, Barry and Tait1990). Furthermore, comparison of 2 T.b. rhodesiense foci in Zambia and Uganda showed that there were 2 genetically distinct groups in the 2 foci (Hide et al. Reference Hide, Buchanan, Welburn, Maudlin, Barry and Tait1991). A detailed analysis of the 1988 sleeping sickness epidemic in Tororo, Uganda, showed that the human isolates (i.e. T.b. rhodesiense) were genetically homogeneous and could be clearly distinguished from non-human isolates (i.e. T.b. brucei) by numerical taxonomy (Hide et al. Reference Hide, Welburn, Tait and Maudlin1994) using RFLP data. Furthermore, the T.b. rhodesiense stocks were found to be frequent in cattle and were closely related to isolates from previous epidemics since the 1960s in that region. Combining molecular and traditional epidemiological data it was shown that humans were 5 times more likely to acquire sleeping sickness via cattle than from other humans (Hide et al. Reference Hide, Tait, Maudlin and Welburn1996), thus demonstrating the importance of cattle as a reservoir for sleeping sickness during an epidemic (Hide, Reference Hide1999). Comparison of epidemic and endemic areas of sleeping sickness (Hide et al. Reference Hide, Angus, Holmes, Maudlin and Welburn1998) and areas free of sleeping sickness (Hide et al. Reference Hide, Tilley, Welburn, Maudlin and Tait2000) revealed the presence of the same human infective strains in patients, cattle and tsetse suggesting that the presence of a human infective strain was not the only factor determining whether an epidemic developed.
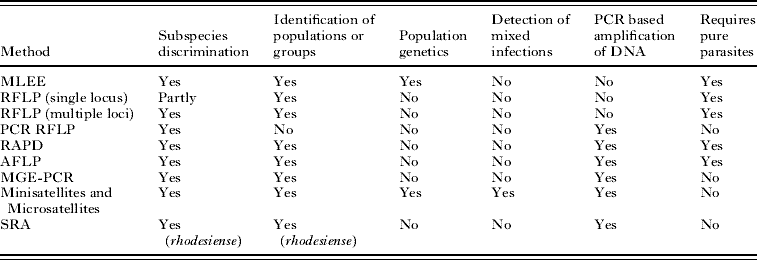
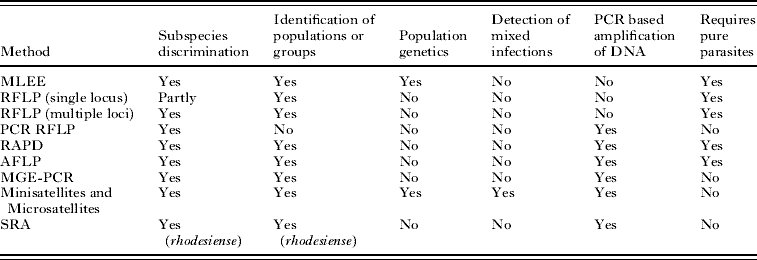
Table 2. Molecular epidemiological methods for analysing African trypanosomes
(The table shows some of the characteristics of each method. (1) Subspecies discrimination, ability to discriminate between the 3 subspecies of Trypanosoma brucei. (2) Identification of populations or groups, the ability to define populations or groups within the subspecies. (3) Population genetics, the ability to be interpreted genetically to enable population genetics to be carried out. (4) Detection of mixed infections, able to detect mixed infections or not to be confounded by the presence of mixed infections. (5) PCR-based amplification of DNA, can be used directly on blood or tissue samples without the need to amplify parasites in experimental animals or in vitro. (6) Requires pure parasites, parasites need to be amplified and purified. MLEE, Multilocus Enzyme Electrophoresis; RFLP, Restriction Fragment Length Polymorphism; PCR-RFLP, PCR amplified RFLP; RAPD, Randomly Amplified Polymorphic DNA; AFLP, Amplified Fragment Length Polymorphism; MGE-PCR, Mobile Genetic Element – PCR; Minisatellites and Microsatellites, analysis of length polymorphism in highly repeated sequences; SRA, detection of the Serum Resistance Associated gene which is found in human infective T.b. rhodesiense.)
It was observed that T.b. rhodesiense isolates circulating in the Tororo region of Uganda were genetically homogeneous whilst the sympatric T.b. brucei isolates were much more diverse (Hide et al. Reference Hide, Welburn, Tait and Maudlin1994). This raised the question as to the contribution of genetic exchange in ‘field’ populations of trypanosomes. High levels of genetic exchange would facilitate the ‘spread’ of phenotypes, such as human infectivity and drug resistance, potentially into new populations and could possibly explain the emergence of new epidemics. This collection of stocks offered an opportunity to investigate this question since they were isolated at the same place and time. However, as suitable DNA-based techniques were not available, it was necessary to resort to MLEE analysis to show that genetic exchange was frequent in T.b. brucei but epidemic in T.b. rhodesiense (Hide et al. Reference Hide, Welburn, Tait and Maudlin1994).
This suggested that genetic exchange could contribute to the genetic variation in trypanosome isolates in the field and importantly could contribute to the epidemiology of the disease. Two problems beset further analysis of these questions. Firstly, the inability to genetically interpret multilocus RFLP markers and, secondly, despite DNA-based methods providing higher levels of resolution, the requirement for sufficient pure parasite material for analysis.
PCR-BASED MOLECULAR EPIDEMIOLOGICAL TOOLS
The advent of PCR to amplify and identify DNA at source or from small amounts of parasite material – such as a single trypanosome (MacLeod et al. Reference MacLeod, Turner and Tait1997; Cox et al. Reference Cox, Tilley, Mcodimba, Fyfe, Hide and Welburn2005) – offered tremendous advantages particularly in the area of diagnosis. In terms of epidemiological analysis, where multilocus analysis was clearly important, this was more difficult. Some single locus typing such as PCR-RFLP was found to be of limited use (Tilley and Hide, Reference Tilley and Hide2001). Random Amplification of Polymorphic DNA (RAPD) analysis was one of the initial solutions to PCR amplification of multiple loci (Mathieu-Daudé et al. Reference Mathieu-Daude, Stevens, Welsh, Tibayrenc and McLelland1995; Stevens et al. Reference Stevens and Tibayrenc1995). This technique relied on the use of a short single PCR primer which randomly annealed to the DNA sample, a banding pattern was produced for each stock and numerical taxonomy used to assess the relatedness of stocks with each other. This was a good multilocus approach, useful with small amounts of DNA, which probably sampled the full extent of the genome rather than being restricted to certain genes or parts of the genome. However, this method is highly susceptible to contaminating host or other DNA – bands will be generated from any DNA template, it is not possible to interpret genetically and is unable to detect mixed infections of trypanosomes. A similar approach, Amplified Fragment Length Polymorphism (AFLP), uses PCR primers based on restriction enzyme sites to look for variation between trypanosome stocks (Agbo et al. Reference Agbo, Majiwa, Claassen and Te Pas2002) and has the same advantages and disadvantages of RAPD analysis. To overcome the problem of contaminating host DNA, a technique termed MGE-PCR (Mobile Genetic Element PCR) was developed utilizing the positional variation of mobile genetic elements to detect genetic variation between parasite isolates (Hide and Tilley, Reference Hide and Tilley2001; Terry et al. Reference Terry, Smith, Duncanson and Hide2001; Tilley et al. Reference Tilley, Welburn, Fevre, Feil and Hide2003). Using a single primer designed to be specific for the repeat regions of the RIME mobile genetic element of T. brucei, amplification of bands between adjacent RIME elements generates a banding pattern which varies from stock to stock based on differences in the positions of RIME elements and so can be used to identify groups of similar isolates by numerical taxonomy. This approach has the advantage that it is PCR-based, specific to T. brucei DNA, multilocus, representative of a significant portion of the genome and generates a single ‘fingerprint’ for each isolate or stock. The disadvantages are that it cannot detect mixed infections and it cannot be interpreted genetically.
The markers that offer the greatest prospects are the mini- and micro-satellite markers (e.g. MacLeod et al. Reference MacLeod, Turner and Tait1997; Biteau et al. Reference Biteau, Bringaud, Gibson, Truc and Baltz2000). These short repeated sequences vary in length or sequence between trypanosome strains and can be amplified and analysed by population genetic methods (MacLeod et al. Reference MacLeod, Turner and Tait2001a). Such markers can be used to detect mixed infections (MacLeod et al. Reference MacLeod, Turner and Tait1999; Koffi et al. Reference Koffi, Solano, Barnabé, De Meeûs, Bucheton, Cuny and Jamonneau2007), are highly specific and sensitive tools for detection of T. brucei ssp. and can be used without the need to amplify parasites in rodents (Morrison et al. Reference Morrison, McCormack, Sweeney, Likeufack, Truc, Turner, Tait and MacLeod2007). The development of PCR-based methods for detailed analysis of trypanosome stocks has opened up many avenues of research. In the remainder of this review we will focus on the contributions of these techniques to our understanding of the diversity of trypanosome isolates in the field, the role of genetic exchange in the epidemiology of sleeping sickness, the nature of human infective strains and the generation of sleeping sickness epidemics.
Many studies have examined the diversity of T. brucei stocks obtained from field isolates. What is clear is that the initial biochemical and molecular studies were correct in suggesting substructuring within each of the subspecies T.b. gambiense and T.b. rhodesiense. Analyses of stocks of T.b. gambiense Type 1 clearly show this group to be genetically highly homogeneous within a focus and distinguishable from T.b. brucei or T.b. rhodesiense by RAPD (Jamonneau et al. Reference Jamonneau, Garcia, Ravel, Cuny, Oury, Solano, N'Guessan, N'Dri, Sanon, Frézil and Truc2002) microsatellite analysis (Biteau et al. Reference Biteau, Bringaud, Gibson, Truc and Baltz2000; Jamonneau et al. Reference Jamonneau, Garcia, Ravel, Cuny, Oury, Solano, N'Guessan, N'Dri, Sanon, Frézil and Truc2002), minisatellite analysis (MacLeod et al. Reference MacLeod, Turner and Tait2001a, Reference MacLeod, Tait and Turnerb, Reference MacLeod, Welburn, Maudlin, Turner and Taitc), MGE-PCR (Simo et al. Reference Simo, Herder, Njiokou, Asonganyi, Tilley and Cuny2005) and AFLP (Simo et al. Reference Simo, Cuny, Demonchy and Herder2008). In most cases, Type 1 T.b. gambiense has been shown to be genetically homogeneous despite differing symptoms in humans being associated with identical parasite genotypes (Jamonneau et al. Reference Jamonneau, Garcia, Ravel, Cuny, Oury, Solano, N'Guessan, N'Dri, Sanon, Frézil and Truc2002). However, more detailed studies have revealed significant differences between isolates from different foci within the Type 1 group (Truc et al. Reference Truc, Ravel, Jamonneau, N'Guessan and Cuny2002; Morrison et al. Reference Morrison, Tait, McCormack, Sweeney, Black, Truc, Likeufack, Turner and MacLeod2008; Koffi et al. Reference Koffi, De Meeûs, Bucheton, Solano, Camara, Kaba, Cuny, Ayala and Jamonneau2009). Interestingly, a new group of T.b. gambiense was identified by micro- and mini-satellites that was associated with asymptomatic disease in humans (Jamonneau et al. Reference Jamonneau, Ravel, Garcia, Koffi, Truc, Laveissière, Herder, Grébaut, Cuny and Solano2004a). It is clear from the microsatellite studies that T.b. gambiense Type 1 is clonal and very limited, if any, genetic exchange occurs in the field (Morrison et al. Reference Morrison, Tait, McCormack, Sweeney, Black, Truc, Likeufack, Turner and MacLeod2008; Koffi et al. Reference Koffi, De Meeûs, Bucheton, Solano, Camara, Kaba, Cuny, Ayala and Jamonneau2009).
The clonal nature of T.b. gambiense has allowed it to be clearly identified and shown to be zoonotic. Early isoenzyme studies demonstrated its presence in dogs, pigs, bovines and wild game (Mehlitz et al. Reference Mehlitz, Zillmann, Scott and Godfrey1982) suggesting a possible animal reservoir. Recent research using PCR-based tools have shown that pigs (Njiokou et al. Reference Njiokou, Laveissière, Simo, Nkinin, Grébaut, Cuny and Herder2006; Simo et al. Reference Simo, Asonganyi, Nkinin, Njiokou and Herder2006) and wild fauna (Njiokou et al. Reference Njiokou, Laveissière, Simo, Nkinin, Grébaut, Cuny and Herder2006) have the potential to be animal reservoirs. Although in another study, using microsatellite analysis, pigs were not considered to be an active reservoir (Jamoneau et al. Reference Jamonneau, Ravel, Koffi, Kaba, Zeze, Ndri, Sane, Coulibaly, Cuny and Solano2004b). It is also possible that untreated, parasitologically unconfirmed, seropositive individuals could also act as a human reservoir (Garcia et al. Reference Garcia, Courtin, Solano, Koffi and Jamonneau2006; Checchi et al. Reference Checchi, Filipe, Barrett and Chandramohan2008). In a clonal pathogen, such as T.b. gambiense, the lack of genetic exchange is helpful since it ensures a reasonable level of stability and reproducibility in genetic markers for use in epidemiological studies.
As with T.b. gambiense, minisatellite analysis has been used to investigate whether T.b. rhodesiense is clonal (MacLeod et al. Reference MacLeod, Tweedie, Welburn, Maudlin, Turner and Tait2000). When the sympatric stocks from Tororo, Uganda (Hide et al. Reference Hide, Welburn, Tait and Maudlin1994), were examined T.b. rhodesiense was found to be clonal, suggesting that little or no genetic exchange is occurring in this population (MacLeod et al. Reference MacLeod, Tweedie, Welburn, Maudlin, Turner and Tait2000). This is in contrast to earlier MLEE studies, using the same stocks, that suggested it was epidemic (Hide et al. Reference Hide, Welburn, Tait and Maudlin1994) – that is, genetic exchange masked by the amplification of a small number of genotypes. This discrepancy might be explained by the more detailed and sophisticated analyses available using minisatellites.
However, as predicted by the earlier MLEE and RFLP studies, genetic differences between different geographical populations can be clearly detected by minisatellite DNA analysis such as between Uganda and Zambia (MacLeod et al. Reference MacLeod, Tait and Turner2001b) and using polymorphism in the SRA (Serum Resistant Associated) gene between populations in Uganda and Malawi or Zambia (Gibson et al. Reference Gibson, Backhouse and Griffiths2002; McLean et al. Reference MacLean, Chisi, Odiit, Gibson, Ferris, Picozzi and Sternberg2004).
The SRA gene (De Greef et al. Reference De Greef, Imberechts, Matthyssens, Van Meirvenne and Hamers1989) has been shown to be capable of transforming a non human-infective T. brucei to a human-infective phenotype (Xong et al. Reference Xong, Vanhamme, Chamekh, Chimfwembe, Van Den Abbeele, Pays, Van Meirvenne, Hamers, De Baetselier and Pays1998). This gene is present in all human-infective field samples from East Africa and absent in animal isolates and has consequently been used as a diagnostic tool for T.b. rhodesiense (Welburn et al. Reference Welburn, Picozzi, Fèvre, Coleman, Odiit, Carrington and Maudlin2001; Gibson et al. Reference Gibson, Backhouse and Griffiths2002). Although this gene looks promising as the candidate gene determining human infectivity in T. brucei, it is clearly absent from both T.b. gambiense Type 1 and 2 (Turner et al. Reference Turner, McLellan, Lindergard, Bisoni, Tait and MacLeod2004) showing that there is more than one mechanism that determines human infectivity. There is growing evidence to suggest that in at least some epidemics, such as the 1998 epidemic in Soroti in Uganda, that the spread of trypanosomes expressing the SRA gene may be responsible for the generation of epidemics (Welburn et al. Reference Welburn, Picozzi, Fèvre, Coleman, Odiit, Carrington and Maudlin2001).
MOLECULAR EPIDEMIOLOGY: THE ORIGINS AND DYNAMICS OF TRYPANOSOMA BRUCEI RHODESIENSE SLEEPING SICKNESS EPIDEMICS
The 1988 Tororo sleeping sickness epidemic in Uganda resulted in a legacy of trypanosome stocks isolated during an epidemic (Hide et al. Reference Hide, Welburn, Tait and Maudlin1994). In addition to analysis by RFLP, they have been analysed using minisatellite markers (MacLeod et al. Reference MacLeod, Tweedie, Welburn, Maudlin, Turner and Tait2000), MGE-PCR (Tilley et al. Reference Tilley, Welburn, Fevre, Feil and Hide2003) and the SRA gene (Welburn et al. Reference Welburn, Picozzi, Fèvre, Coleman, Odiit, Carrington and Maudlin2001). A clear distinction has emerged between the human infective stocks and non human-infective stocks in this region. The finding that cattle are an important reservoir for human infective stocks (Hide et al. Reference Hide, Tait, Maudlin and Welburn1996) has been confirmed by more recent studies (Welburn et al. Reference Welburn, Picozzi, Fèvre, Coleman, Odiit, Carrington and Maudlin2001). Isolates collected from humans 10 years on from this epidemic have been shown by MGE-PCR to contain some identical genotypes thus confirming the stability of genotypes within this focus (Tilley et al. Reference Tilley, Welburn, Fevre, Feil and Hide2003). A new epidemic started in December 1998 in Soroti, Uganda (Fèvre et al. Reference Fèvre, Coleman, Odiit, Magona, Welburn and Woolhouse2001), and is currently ongoing and spreading north and west (Fèvre et al. Reference Fèvre, Picozzi, Fyfe, Waiswa, Odiit, Coleman and Welburn2005). Based on the evidence that cattle are an important reservoir for sleeping sickness, it has been postulated that cattle infected with human-infective T. brucei were imported from the Tororo District into Soroti (Fèvre et al. Reference Fèvre, Coleman, Odiit, Magona, Welburn and Woolhouse2001). These cattle were then fed upon by local tsetse, passed onto the local people and the epidemic started. Statistical analysis of the location of sleeping sickness cases in relation to the Soroti cattle market (Fèvre et al. Reference Fèvre, Coleman, Odiit, Magona, Welburn and Woolhouse2001) and the presence of SRA positive T. brucei isolated from the cattle at this market has supported this view (Welburn et al. Reference Welburn, Picozzi, Fèvre, Coleman, Odiit, Carrington and Maudlin2001). Other studies conducted on T. brucei isolates collected from Soroti suggest that they have a different clinical disease profile (e.g. more chronic presentation) to isolates collected in Tororo and also the corresponding microsatellite profiles were found to be different (MacLean et al. Reference MacLean, Odiit, MacLeod, Morrison, Sweeney, Cooper, Kennedy and Sternberg2007). This supports the view that, although some of the isolates have probably originated in Tororo, there may be other origins of these T. brucei isolates. These results clearly show that the importation of cattle from a high risk area may have been responsible for the generation of this new epidemic. It also shows the importance of tracking cattle movements particularly from high risk areas, such as Tororo, and the identification of the geographical locations of high risk areas.
Based on the extensive molecular epidemiological data from Uganda, it is possible to formulate a model of how sleeping sickness foci are generated and how epidemics develop. It is clear from the data that foci, such as the Busoga focus, must have the presence of a human-infective T. brucei strain (‘T.b. rhodesiense’). Molecular epidemiological studies suggest that this may be a single predominant clonal strain which remains stable over time (although multiple clonal strains could also be present). New epidemics can be generated by the influx of a human strain into an area previously occupied by only animal strains. This could be by the importation of a reservoir species, such as cattle, infected with a human strain. Local socio-political customs or events may determine the amplitude and duration of the epidemic. Despite having a wide antigenic repertoire, clonality will ensure that any given T.b. rhodesiense strain will have reduced genetic variability. Consequently, with time, a local human population will become immune/less susceptible or subjected to health interventions causing the epidemic to decline. On entering a naïve population, such as Soroti, clonal human strains would spread quickly. It is clear that different foci appear to have genetically different human strains (T.b. rhodesiense or T.b. gambiense) which could be generated either by the SRA gene being introduced into a new genetic background or by the introduction of one of the other mechanisms of human infectivity emerging in a previously non human-infective strain. This mechanism could involve genetic exchange between a non human- and a human-infective strain. With time and selection through humans, emergent human strains might become established and generate new foci. The data suggest that, in the field, genetic exchange occurs but is masked by the epidemic spread of a small number of strains. The relatively small numbers of human infective strains, the relatively small number of foci and the long-term stability of foci might suggest that there is a minimal effect of genetic exchange in generating new human strains and foci. However, in practice, the generation of even a single new focus creates a significant health issue. Important questions arise from this model. Are there low frequencies of genetic exchange generating new human-infective strains or is the generation of new human-infective strains by genetic exchange frequent but fixation of those strains within the population (to produce a focus) infrequent? Also, given the relatively limited data from foci in Zambia, Tanzania, Malawi and Mozambique, are the conclusions from the Ugandan studies typical for T.b. rhodesiense? As can be seen from this review, the molecular tools are now available to address such questions.
As with any model, this one generates more questions than it answers. We have come a long way to understanding the generation of sleeping sickness epidemics but further questions need to be addressed. The future of molecular epidemiology will be based on our increasing ability to be able to conduct large-scale epidemiological studies which will involve carefully controlled sampling strategies, application of high throughput whole genome technologies for both host and parasite, development of Single Nucleotide Polymorphism (SNP) chips, development of bioinformatics tools and integration with mathematical models of disease transmission. These approaches and technologies provide opportunities for the future and offer the prospect of addressing some of the questions raised in this review.
We would like to acknowledge all of our colleagues who have contributed to the studies we have reviewed here. While we have tried to provide appropriate coverage in this review, we acknowledge that a great deal of important work could not be included here for reasons of space.