INTRODUCTION
Lymphatic filariasis infection caused by Wuchereria bancrofti, Brugia malayi and Brugia timori affects 1·3 billion people living in many endemic countries (World Health Organization, 2009; Bockarie and Deb, Reference Bockarie and Deb2010). Although significant advances have been made in parasitic research, little progress has been made in the chemotherapy of lymphatic filariasis. The treatment of the filariasis with the currently available drugs is highly unsatisfactory. Diethylcarbamazine (DEC), ivermectin alone and in combination with albendazole are presently being used to control the filarial infection (Critchley et al. Reference Critchley, Addiss, Ejere, Gamble, Garner and Gelband2005; Ramzy et al. Reference Ramzy, El Setouhy, Helmy, Ahmed, Abd Elaziz, Farid, Shannon and Weil2006). These drugs are basically microfilaricidal in nature and exert little effect on adult worms, thus repeated treatment is required for interrupting the transmission of the filarial infection. Indications of resistance have been reported for albendazole and ivermectin (Osei-Atweneboana et al. Reference Osei-Atweneboana, Eng, Boakye, Gyapong and Prichard2007; Schwab et al. Reference Schwab, Churcher, Schwab, Basanez and Prichard2007). Efforts are now being made to identify macrofilaricides/embryostatic agents with microfilaricidal activity to reduce/eliminate the problem of emerging drug resistance. For drug discovery against any parasitic diseases, identification and characterization of the enzymes that play crucial roles in the growth and survival of the parasites is needed. The availability of the complete genome information for B. malayi will help identification of the key enzyme that can be utilized as a drug target. The enzymes of the pentose phosphate pathway (PPP), an alternative route to glycolysis for degradation of glucose (Winiarska et al. Reference Winiarska, Drozak, Wegrzynowicz, Jagielski and Bryla2003), in nematodes can serve as an attractive drug target. Glucose-6-phosphate dehydrogenase (G6PD) (EC1.1.1.49), a rate-limiting enzyme of PPP plays a crucial role in maintaining the NADPH/NADP+, GSH/GSSG ratio, protection from redox stress-induced apoptosis and in cell growth. The tightly controlled concentrations of ROS and fluctuations in redox potential are important mediators of signalling processes, the stress response and development and ageing (Saxena et al. Reference Saxena, Pandya, Gupta and Shukla1996; Schulz et al. Reference Schulz, Zarse, Voigt, Urban, Birringer and Ristow2007; Van Raamsdonk and Hekimi, Reference Van Raamsdonk and Hekimi2010). Inhibition of G6PD activity causes accumulation of 6-phosphogluconate, which inhibits glycolysis by competitively inhibiting glucose-6-phosphate-isomerase (Cordeiro et al. Reference Cordeiro, Thiemann and Michels2009). Therefore a decrease in G6PD activity and as a result NADPH level will impair the entire antioxidant system (Tian et al. Reference Tian, Braunstein, Apse, Pang, Rose, Tian and Stanton1999; Ciftci et al. Reference Ciftci, Beydemir, Yilmaz and Altikat2003; Filosa et al. Reference Filosa, Fico, Paglialunga, Balestrieri, Crooke, Verde, Abrescia, Bautista and Martini2003; Fico et al. Reference Fico, Paglialunga, Cigliano, Abrescia, Verde, Martini, Iaccarino and Filosa2004; Zhang et al. Reference Zhang, Liew, Handy, Zhang, Leopold, Hu, Lili Guo, Kulkarni, Loscalzo and Stanton2010). Cordeiro et al. (Reference Cordeiro, Thiemann and Michels2009) have reported that G6PD plays an important role in growth and survival of the bloodstream-form trypanosomes. RNAi-mediated reduction of G6PD levels in Trypanosoma brucei bloodstream forms validated this enzyme as a drug target against human trypanosomiasis. Gupta et al. (Reference Gupta, Igoillo-Esteve, Michels and Cordeiro2011) reported that Trypanosoma G6PD plays a prominent role in the defence of the parasite against oxidative stress and is an important drug target for chemotherapy. The present communication reports the cloning, expression and kinetic characterization of the recombinant G6PD of B. malayi (rBmG6PD) with the aim of understanding the differences between the host and parasitic enzyme which can be fruitfully utilized for designing of selective inhibitors with antifilarial activity.
MATERIALS AND METHODS
All the reagents were procured from Sigma (St Louis, MO, USA). IPTG, pre-stained markers and restriction enzymes BamHI and EcoRI were purchased from MBI Fermentas (Hanover, Maryland, USA). The DNA marker was procured from Bangalore Genei, India.
cDNA synthesis and PCR amplification
Total RNA of B. malayi was isolated using the TRIzol reagent (Invitrogen) and synthesis of cDNA was carried out using cDNA synthesis kit (Invitrogen, USA). The putative BmG6PD gene was PCR amplified from the cDNA of the parasite using gene-specific forward primers 5′ GGATCCGATGTCGCATGAAAATTCGCCGA 3′ having the BamHI restriction site (underlined) and the reverse primer 5′ GAATTCACGTTCAGTGTTTGGAATCCATTTG 3′ having the EcoRI restriction site (underlined) designed on the basis of sequence information available at www.kegg.jp.
Cloning and expression of BmG6PD
The PCR-amplified fragment was gel purified and cloned into pGEMT Easy cloning vector as reported earlier (Singh et al. Reference Singh, Joshi, Arya, Kayastha, Srivastava, Tripathi and Saxena2008). The gene was digested from the plasmid pGEMT-BmG6PD by restriction endonucleases BamHI and EcoRI and subcloned into pTriEx-4 vector resulting in plasmid pTriEx-BmG6PD. This plasmid was transformed into Escherichia coli DH5α cells. Transformants were screened through colony PCR and restriction digestion. Cloning of BmG6PD was finally confirmed by sequencing of the clone.
To study the expression of recombinant BmG6PD, plasmid pTriEx-BmG6PD was transformed in E. coli C41 cells. A single colony from the transformed plate was inoculated in 5 mL of Luria-Bertani (LB) broth containing 100 μg mL−1 ampicillin. Cells were grown for 14–16 h at 37 °C with shaking at 180 rpm. Then 1·0% (v/v) of the overnight grown culture was inoculated into 500 mL of fresh LB containing 100 μg mL−1 ampicillin. Cultures were grown at 37 °C until an optical density 0·4–0·6 at 600 nm (OD600) was reached; at this stage the culture was induced with 0·8 mm isopropyl-β-thiogalactopyranoside (IPTG). Cultures were then grown at 20 °C for 14–16 h. Cells were harvested by centrifugation at 8000 g for 10 min at 4 °C. The cell pellet was stored at −20 °C until used.
Purification of rBmG6PD
rBmG6PD having a 6x-His tag attached to its N and C terminals was over-expressed by inducing E. coli C-41 cells with IPTG (0·8 mm) and affinity purified by Ni2+-nitrilotriacetic acid resin (Ni2+-NTA). The cells were lysed by pulse sonication in lysis buffer (50 mm NaH2PO4, 600 mm NaCl, 10 mm imidazole, 10% (v/v) glycerol) containing a protease inhibitor cocktail (Sigma). The supernatant obtained after centrifugation at 10 000 g for 45 min was loaded onto a Ni2+-NTA column pre-equilibrated with lysis buffer and contaminating proteins were removed by washing with the same buffer containing 50 mm imidazole, 0·1% (v/v) Triton X-100 and 1 mmβ-mercaptoethanol. Recombinant protein was eluted in fractions of 1·0 mL using 12 mL of lysis buffer containing 300 mm NaCl and 250 mm imidazole. The eluted recombinant protein was concentrated by centriprep (Milipore) and the protein concentration was determined using the method of Bradford (Reference Bradford1976). The purification was performed at 4 °C and the recombinant BmG6PD was analysed by SDS–PAGE followed by R-250 Coomassie blue staining. Western blotting using anti-His antibodies was employed to check the purity of the eluted protein.
Determination of native molecular weight
The subunit mass of rBmG6PD was determined according to the method of Laemmli (Reference Laemmli1970). The native molecular mass of the enzyme was determined with the help of a calibration curve between elution volume and log molecular mass (kDa) of standard marker proteins by the Superdex 200 HR 10/300 column (manufacturer's exclusion limit 600 kDa for proteins) on AKTA FPLC (Amersham Pharmacia Biotech, Sweden). The protein was eluted with 50 mm sodium phosphate buffer (pH 7·0) containing 100 mm NaCl at a flow rate of 0·5 mL min−1. Ovalbumin (42·7 kDa), albumin (67 kDa), aldolase (158 kDa) and ApoFerritin (443 kDa) were used as standards. The native molecular weight of rBmG6PD was further confirmed by cross-linking with 1·0% (v/v) glutaraldehyde.
Generation of polyclonal antibodies and Western immunoblotting
For the generation of polyclonal antibodies, 250 μg purified rBmG6PD protein was emulsified with an equal volume of Freund's complete adjuvant (Sigma, USA) and injected subcutaneously in a 6-month-old rabbit. Two booster doses of 150 μg rBmG6PD, in Freund's incomplete adjuvant (Sigma, USA), were given after the 3rd and 5th weeks. The rabbit serum was collected after 7 days of the second booster dose and the antibody titre was measured by ELISA. Anti-BmG6PD antibodies were used in Western blot to determine whether the corresponding native protein was present in microfilariae/adult B. malayi. Samples of 50 μg of parasite protein as well as recombinant protein were resolved on 10% (w/v) SDS–PAGE gel and electro-blotted onto the nitrocellulose membrane (Sambrook et al. Reference Sambrook, Fritsch and Maniatis1989). The membrane was probed with anti-BmG6PD serum (1:10 000 dilution) as the primary antibody, followed by HRP-conjugated anti-rabbit IgG as the secondary antibody. The blot was developed by chromogenic peroxidase reaction with 3,3′-diaminobenzidine (Singh et al. Reference Singh, Joshi, Arya, Kayastha, Srivastava, Tripathi and Saxena2008).
Enzyme assay and kinetic investigations
Enzyme activity was determined spectrophotometrically, by monitoring the NADPH production at 340 nm according to Betke et al. (Reference Betke, Beutler, Brewer, Kirkman, Luzzatto, Motulsky, Ramot and Siniscalco1967). The assay mixture contained 10 mm MgCl2, 0·2 mm NADP+ and 0·6 mm glucose-6-phosphate (G6P), 100 mm Tris–HCl buffer, pH 8·0. The optimum pH was determined by measuring activity with potassium phosphate buffer, pH 6·5–8·0, Tris–HCl buffer, pH 8·0–9·0 and glycine-NaOH buffer, pH 9·0–10·5. The effect of metal ions and various inhibitors, namely dihydroepiandroesterone (DHEA), 6-aminonicotinamide (6-AN), p-chloromercuribenzoate (pCMB), N-ethylmaleimide (NEM) and metal chelator-like ethylenediaminetetraacetic acid (EDTA) was studied by incubating the enzyme with inhibitor for 10 min at 25 °C. The inhibition of BmG6PD by NADPH, nucleotides: adenosine triphosphate (ATP), adenosine diphosphate (ADP), adenosine monophosphate (AMP), adenosine 5′ [γ-thio] triphosphate (ATP-γ-S), adenosine 5′ [β,γ-imido] triphosphate (ATP-β,γ-NH), adenosine 5′ [β-thio] diphosphate (ADP-β-S), regulatory ligands: phosphoenolpyruvate and fructose-6-phosphate was determined by incubating the enzyme for 10 min at room temperature and starting the reaction by addition of substrate. The number of SH groups in rBmG6PD was estimated by studying its reaction with 5, 5′ dithiobis-(2-nitrobenzoate) DTNB. The appearance of the yellow-coloured product 2-nitromercaptobenzoate was monitored at 415 nm. The effects of known antifilarials and CDRI synthesized compounds on rBmG6PD were also studied in similar manner.
GdmCl/urea denaturation of rBmG6PD
Denaturation of rBmG6PD by GdmCl/urea was carried out by incubating 5 μ m of rBmG6PD in 50 mm sodium phosphate buffer (pH 7·0) with increasing concentrations of guanidine hydrochloride (GdmCl)/urea at 4 °C for 4 h. The effects of denaturants on fluorescence spectra of rBmG6PD were recorded in a Varian fluorescence spectrophotometer. The excitation wavelength for tryptophan was 295 nm and the emission was recorded from 300 to 400 nm. Circular dichroism measurements were made with a Jasco J810 spectropolarimeter calibrated with ammonium (+)-10-camphorsulfonate. The results are expressed as the mean residual ellipticity (θ), which is defined as (θ)=100×θobs/(lc), where θobs is the observed ellipticity in degrees, c is the concentration in mol residue per litre, and l is the length of the light path in centimetres. The CD spectra were measured at an enzyme concentration of 5 μ m with a 1 mm cell at 25 °C. The values obtained were normalized by subtracting the baseline recorded for buffer having the same concentration of denaturants under similar conditions.
RESULTS
Expression and purification of rBmG6PD
Brugia malayi G6PD was successfully cloned, expressed and purified by Ni2+-NTA affinity chromatography. The protein was eluted from the affinity column by 250 mm imidazole (Supplementary Fig. S1A, Online version only). The expressed protein was also characterized using anti-His antibodies (Supplementary Fig. S1B, Online version only). The subunit molecular weight of recombinant protein was found to be ∼75 kDa, along with the vector encoding a molecular weight of 10 kDa.
Native molecular weight
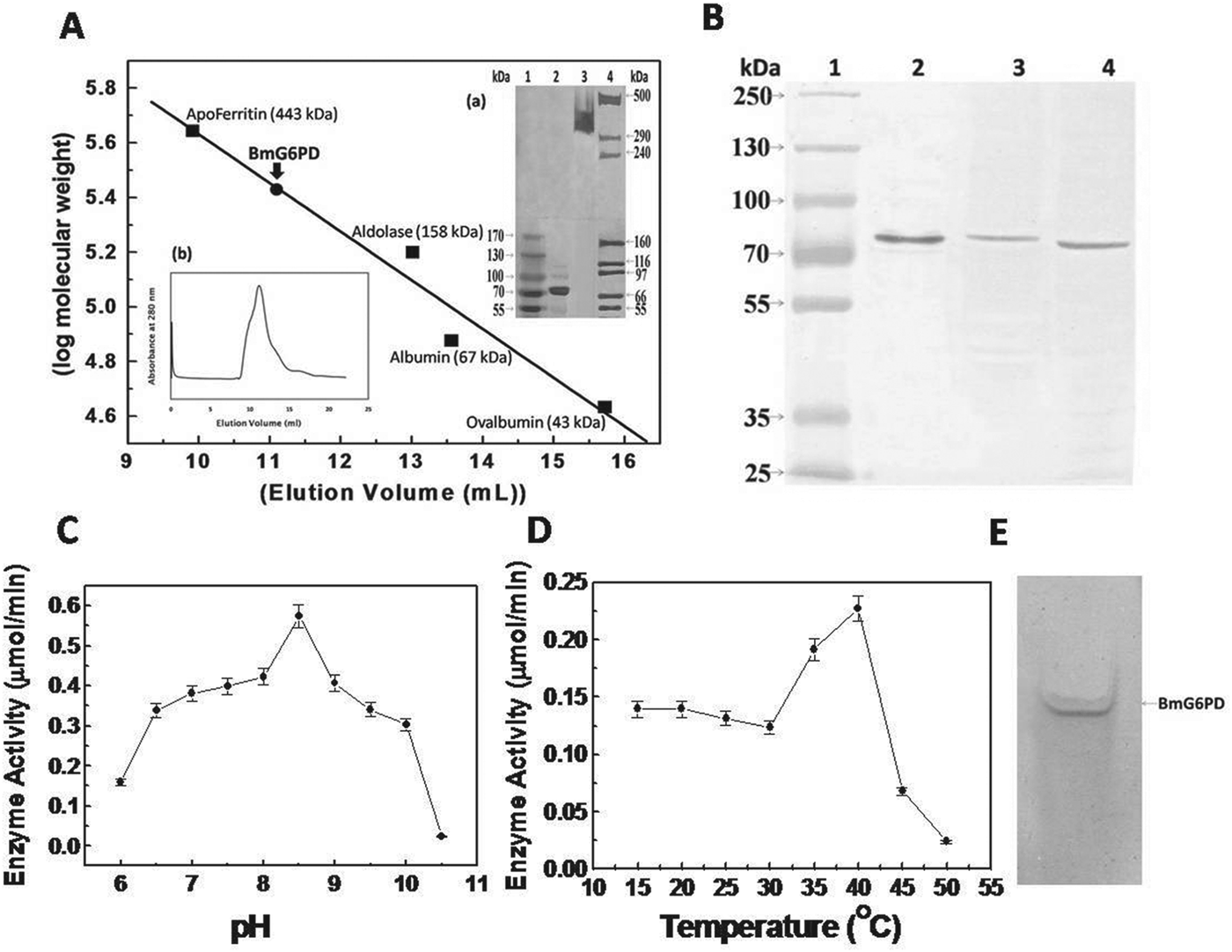
The native molecular weight of rBmG6PD was ∼300 kDa as obtained by size-exclusion chromatography, indicating that rBmG6PD is a tetramer of ∼75 kDa subunits (Fig. 1A). The cross-linking of the monomeric subunits using glutaraldehyde as cross-linker and analysis on a 5–8% SDS–PAGE gradient gel showed similar results (Fig. 1A inset a).

Fig. 1. (A) Determination of native molecular mass of rBmG6PD by FPLC using Tricorn Superdex-200 gel filtration column. The column was calibrated with standard protein markers: Inset shows (a) Glutaraldehyde cross-linked rBmG6PD on 5–8% SDS–PAGE gel. Lane 1, pre-stained protein ladder; lane 2, purified rBmG6PD; lane 3, cross-linked rBmG6PD; lane 4, high molecular weight protein ladder, (b) Size Exclusion Chromatography profile of rBmG6PD with absorbance at 280 nm. (B) Detection of BmG6PD in microfilariae and adults of Brugia malayi with antiserum raised against recombinant BmG6PD. Lane 1, pre-stained protein ladder; lane 2, recombinant BmG6PD; lane 3, B. malayi microfilariae extract; lane 4, B. malayi adult extract. (C) Effect of pH on rBmG6PD activity. (D) Effect of temperature on rBmG6PD activity. (E) Zymogram analysis of BmG6PD.
Specificity of anti-BmG6PD antibodies and Western immunoblotting
The titre of anti-BmG6PD antibody raised against recombinant protein was found to be 1:125 000. For Western immunoblotting, Fig. 1B illustrates the specificity of the antibody for the purified recombinant BmG6PD as well as the presence of G6PD protein in microfilariae and adult stages of the parasite.
Kinetic studies of rBmG6PD
The catalytic activity of rBmG6PD increased with an increase in pH and showed maximum activity at pH 8·5 (Fig. 1C); the optimum temperature was observed to be 40 °C (Fig. 1D) with complete loss of activity at 50 °C. The enzyme followed Michaelis–Menten kinetics with respect to G6P and NADP+ and a Lineweaver–Burk plot was used for the evaluation of km values, which were 0·245±0·008 mm for G6P and 0·014±0·0003 mm for NADP+, in addition to Kcat which was 40 s−1. The specific activity of the rBmG6PD was 0·535 Unit mg−1 protein. To further validate the G6PD activity of recombinant BmG6PD, zymogram analysis was performed (Singh et al. Reference Singh, Joshi, Arya, Kayastha, Srivastava, Tripathi and Saxena2008). A blue band was observed in the gel due to formation of formazan that precipitated in the gel at the site of G6PD activity (Fig. 1E).
Effect of metabolite, nucleotides and regulatory ligands
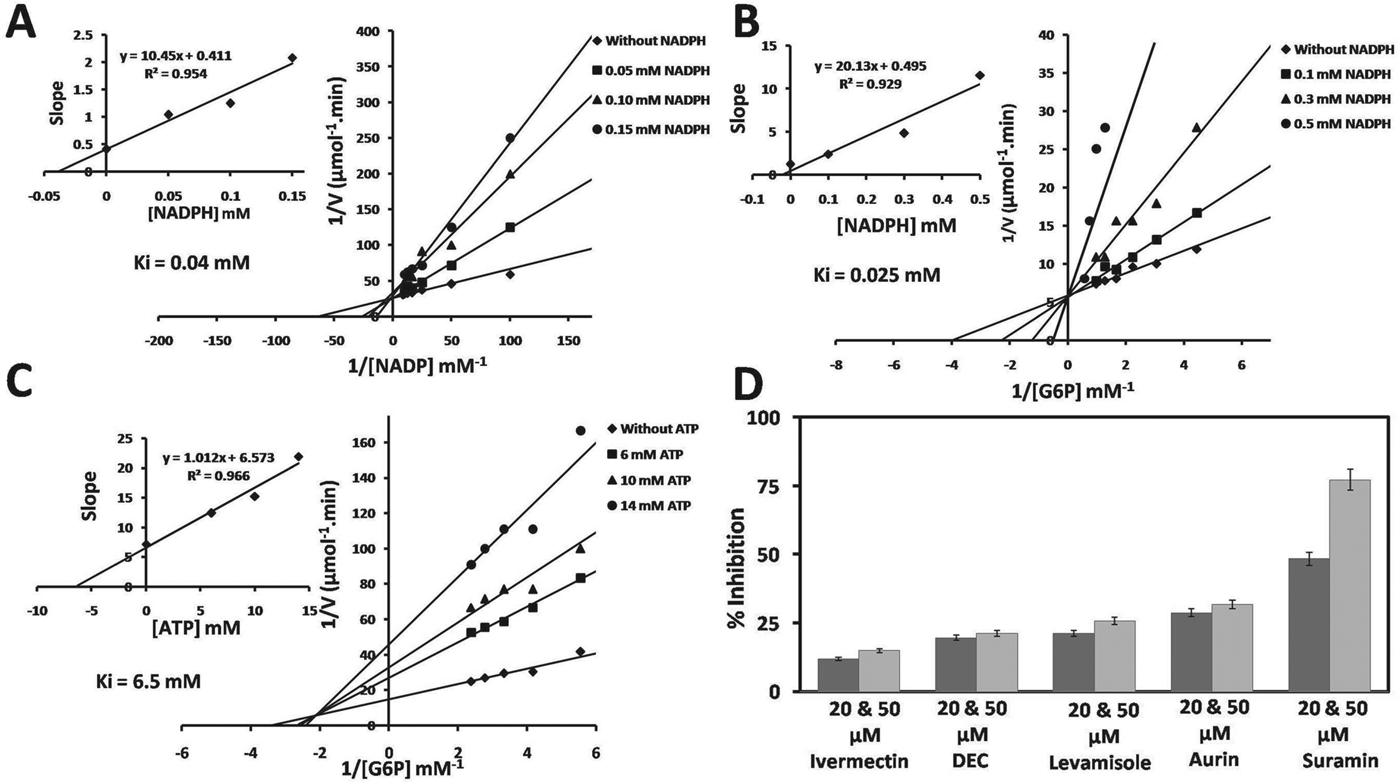
NADP-linked rBmG6PD enzyme activity was competitively inhibited by NADPH with respect to NADP+ and G6P, along with an inhibition constant (Ki) value of 0·04 and 0·025 mm for NADP+ and G6P respectively (Fig. 2A and B). NADP-specific rBmG6PD activity was 31% inhibited by 5 mm ATP while ADP and AMP showed no significant changes in the rBmG6PD activity. Non-hydrolysable ATP and ADP analogues, namely ATP-γ-S, ATP-β,γ-NH and ADP-β-S inhibited rBmG6PD activity to a different extent (Table 1) and10 mm phosphoenol pyruvate (PEP) showed 25% reduction in activity. ATP inhibited the rBmG6PD in a non-competitive manner with respect to G6P, with a Ki value of 6·5 mm (Fig. 2C). Table 2 shows the inhibition of rBmG6PD by CDRI synthesized compounds. The antifilarial suramin inhibited rBmG6PD by ∼77% at 50 μ m concentration, while ivermectin, DEC, levamisole and aurin exhibited no significant effect (Fig. 2D).
Fig. 2. Lineweaver-Burk plot with different concentrations of substrate. (A) Varying NADP concentrations with 3 fixed concentrations of NADPH. (B) Varying G6P concentrations with 3 fixed concentrations of NADPH. (C) Varying G6P concentrations with 3 fixed concentrations of ATP. (D) Effect of antifilarial compounds on rBmG6PD. The enzyme was incubated with antifilarial for 10 min before measuring activity. Results expressed as per cent inhibition of rBmG6PD activity with respect to control.
Table 1. Effect of various ligands on recombinant Brugia malayi G6PD

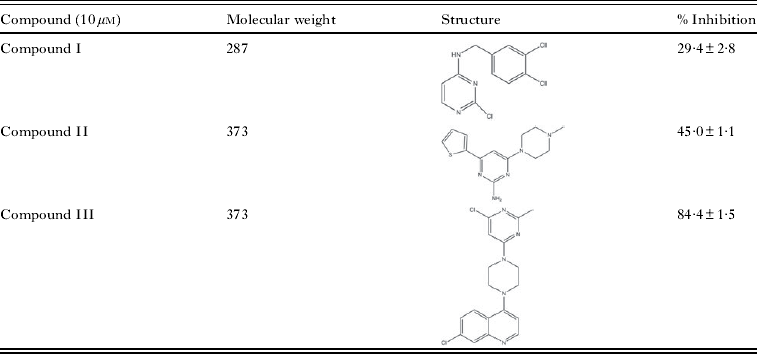
Table 2. Inhibition of rBmG6PD by CDRI compounds
Estimation and significance of SH groups for enzyme activity
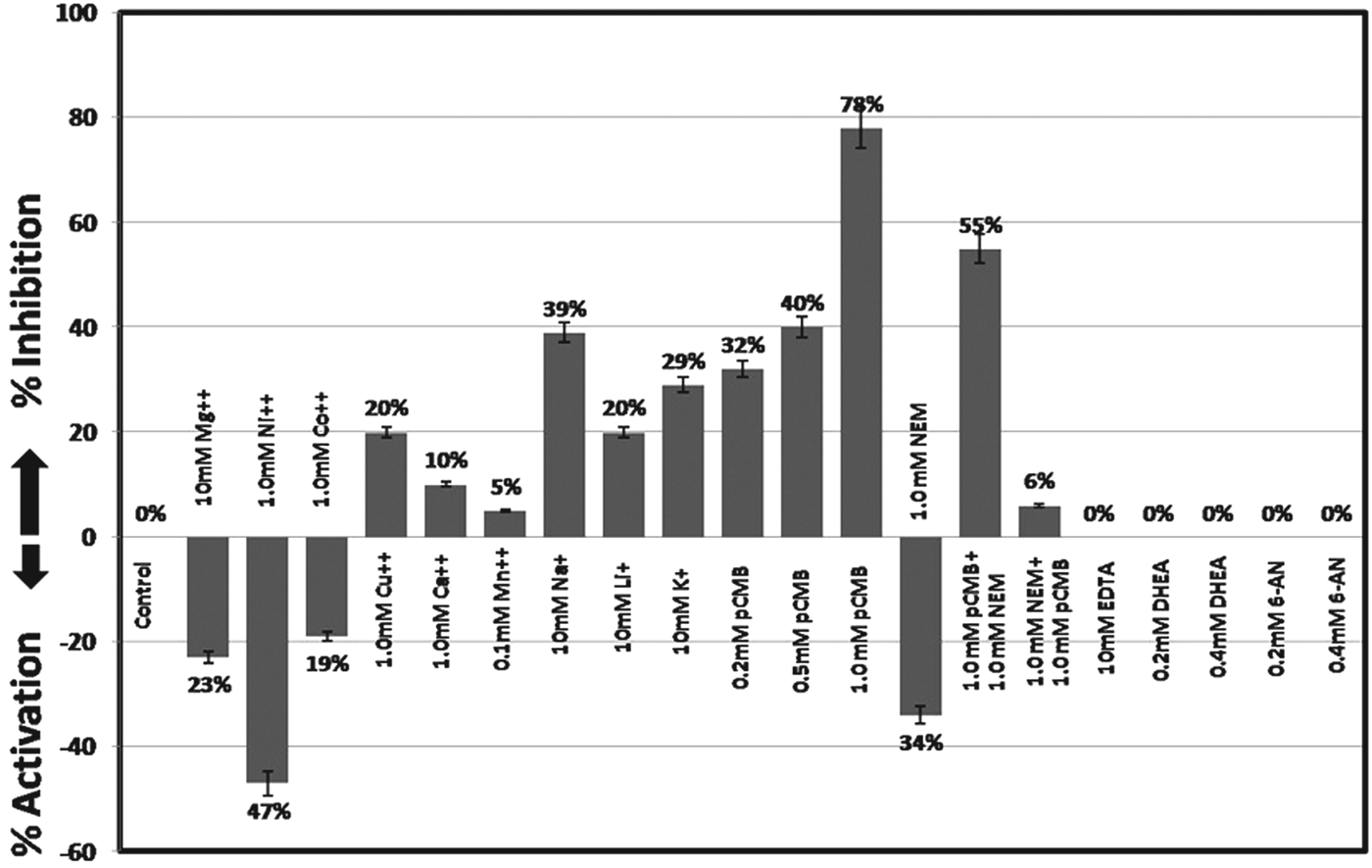
rBmG6PD contained 0·27 mol of SH groups per mol of monomer under denatured conditions and 0·02 mol of reactive SH groups per mol of monomer under native conditions. The SH group inhibitors, pCMB and NEM significantly affected the enzyme, indicating that SH groups play an important role in activity (Fig. 3).
Fig. 3. Effect of metal ions and inhibitors on the activity of rBmG6PD. The values are the mean of 3 different experiments. The enzyme was incubated with the inhibitor for 10 min and the reaction was started by addition of the substrate.
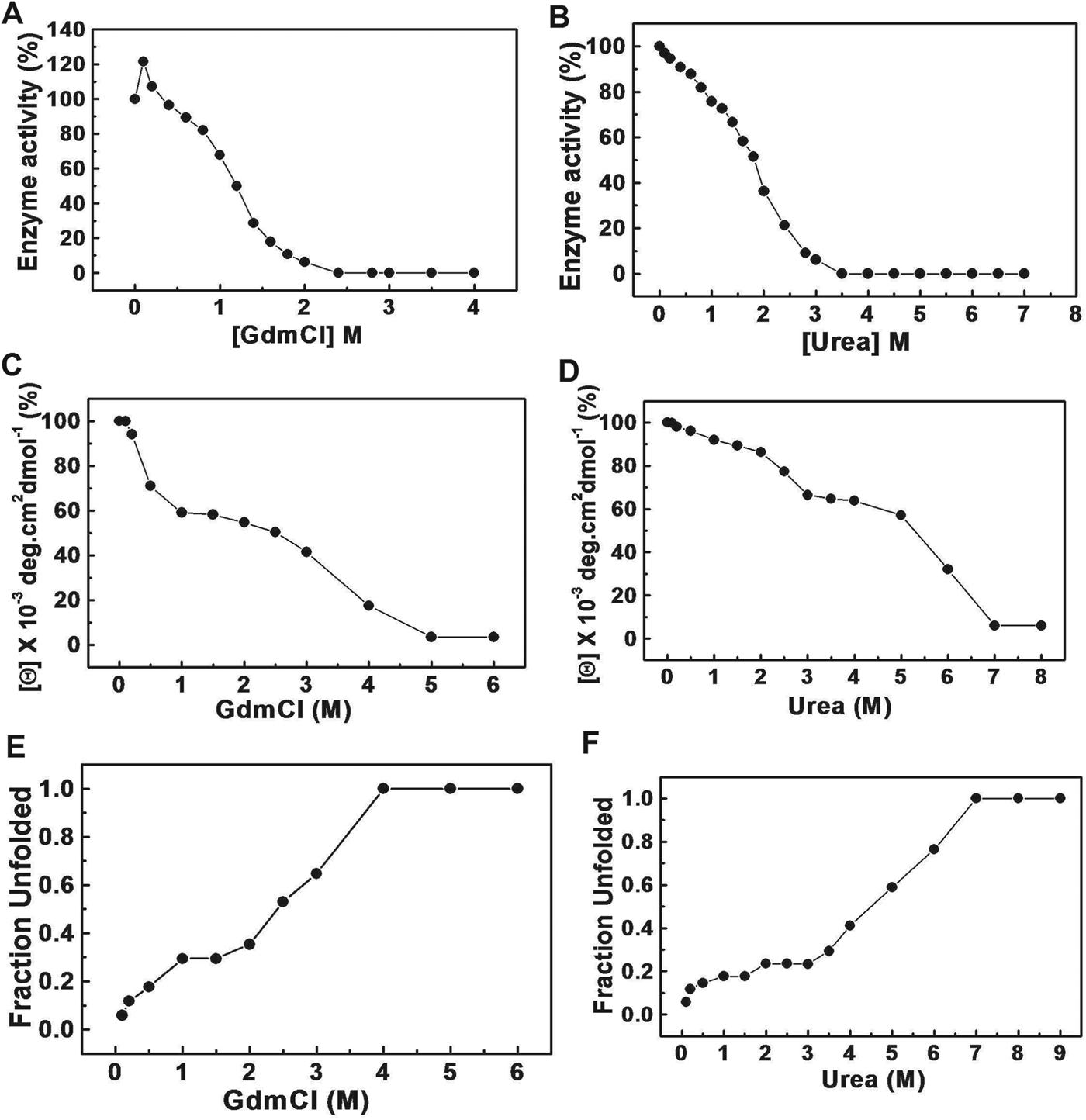
Effect of GdmCl/urea on the activity
As shown in Fig. 4A, rBmG6PD was activated by low concentrations (0·2 m) of GdmCl and the activity gradually decreased with a further increase in GdmCl concentration up to 0·75 m. However, a sharp loss of enzymatic activity above 0·75 m GdmCl was obtained and at 2 m GdmCl complete inactivation of enzyme activity was observed. A gradual decrease in enzyme activity was observed up to 3·0 m urea concentration and 100% inhibition was observed above 3·0 m urea concentration (Fig. 4B).
Fig. 4. Effect of GdmCl and urea on rBmG6PD. (A) Changes in enzyme activity upon incubation with increasing concentrations of GdmCl. (B) Changes in rBmG6PD activity with increasing concentrations of urea. The enzyme was incubated with the desired concentration of GdmCl/urea for 4 h at 4 °C and activity was measured by the method described in the Materials and Methods section. Values are plotted as the remaining enzymatic activity in the presence of different concentrations of GdmCl/urea. (C) GdmCl-induced denaturation of rBmG6PD as monitored by following changes in ellipticity at 222 nm measured by the far-UV CD spectra. (D) Urea-induced changes in ellipticity at 222 nm obtained from the far-UV CD spectra of rBmG6PD. (E) GdmCl-induced changes in tryptophan fluorescence of rBmG6PD. (F) Urea-induced tryptophan fluorescence spectra of rBmG6PD.
GdmCl/urea-induced unfolding of rBmG6PD
Fluorescence spectra provide information on the structural and dynamic properties of the protein. CD analysis of rBmG6PD was performed to determine the structural integrity and helix content. Analysis of the CD spectrum suggested that rBmG6PD contained 37% α-helix and 26% β-sheets while the remaining structure is a randomly coiled structure (data not shown). GdmCl/urea-induced alterations in the tertiary and secondary structures of the recombinant protein, were monitored by studying the tryptophan fluorescence and far-UV CD measurement respectively. GdmCl/urea-induced denaturation of the enzyme showed an apparent three-state unfolding transition. Figure 4 shows the effect of increasing GdmCl/urea concentrations on rBmG6PD by CD ellipticity at 222 nm (Fig. 4C and D) and tryptophan fluorescence (Fig. 4E and F).
DISCUSSION
G6PD is a NADP+-dependent first rate-limiting enzyme of the PPP which produces pentoses and NADPH. The activities of G6PD and 6-phosphogluconate dehydrogenase depend on the energy and redox potential of the cell (Igoillo-Esteve and Cazzulo, Reference Igoillo-Esteve and Cazzulo2006). They are repressed by ATP, NADH and NADPH and control the expression of the genes of the defined PPP (Ibraheem et al. Reference Ibraheem, Adewale and Afolayan2005).
The characterization of B. malayi, a human filarial parasite, G6PD showed that the aberrant mobility of the protein on SDS–PAGE can mainly be accounted for by the properties of the rBmG6PD protein; however, the open reading frame (ORF) of the BmG6PD cDNA encodes only a 58 kDa protein. Thus the difference between the theoretical molecular weights and the apparent molecular weights of the BmG6PD may be due to its negative charge (15% acidic amino acids), a finding that is quite common and has been observed with other proteins (Klenova et al. Reference Klenova, Nicolas, Sally, Carne, Lee, Lobanenkov and Goodwin1997). The Km value for NADP+ is lower than G6P, indicating that rBmG6PD shows higher affinity towards NADP+ as compared with G6P substrate. BmG6PD has high affinity for the G6P as compared with human G6PD (Bautista et al. Reference Bautista, Manson and Luzzatto1992). However, the affinity for NADP was higher in filarial and other parasites as compared with human. G6PD has been purified from Trypanosoma cruzi (Igoillo-Esteve and Cazzulo, Reference Igoillo-Esteve and Cazzulo2006) and the authors demonstrated that the Km for G6P and NADP in the recombinant G6PD was lower as compared with the partially purified enzyme (288 μ m). The apparent Km from NADP was similar for recombinant protein and the native protein. The molecular weight of rBmG6PD determined by FPLC and glutaraldehyde cross-linking indicated its tetrameric nature, which was stable at low pH and resembled human G6PD (Wrigley et al. Reference Wrigley, Heather, Bonsignore and De Flore1972). Parasitic G6PD was degraded to dimer and monomer forms at higher pH, which are catalytically inactive, whereas the dimer form of human G6PD has been shown to be catalytically active (Wang and Engel, Reference Wang and Engel2009).
NADPH and ATP showed competitive and non-competitive inhibition of rBmG6PD respectively, but the Ki values were higher as compared with other parasites. rBmG6PD activity was not strongly inhibited by NADPH as compared with human and other parasites, indicating that the enzymatic activity is regulated by the NADP+/NADPH ratio in the cytosol as reported earlier (Ciftci et al. Reference Ciftci, Beydemir, Yilmaz and Altikat2003; Ibraheem et al. Reference Ibraheem, Adewale and Afolayan2005). The activity of rBmG6PD is strongly influenced by nucleotides suggesting their regulatory role. Variability in ATP concentration inhibited human G6PD while no inhibition was observed for sheep G6PD (Smith and Anwer, Reference Smith and Anwer1970). rBmG6PD was not strongly inhibited by ADP and AMP whereas ATP, ATP-γ-S, ATP-β,γ-NH and ADP-β-S showed significant inhibition. PEP has been reported to be a potent inhibitor of bacterial G6PDs (Ibraheem et al. Reference Ibraheem, Adewale and Afolayan2005), nevertheless the allostearic effect of PEP on BmG6PD was less prominent, and PEP is probably not a significant effector of this enzyme under physiological conditions. On the other hand, PEP has been shown to be a general regulator for G6PD in some of the organisms which depend on this enzyme for hexose catabolism (Ibraheem et al. Reference Ibraheem, Adewale and Afolayan2005). rBmG6PD was not inhibited by EDTA indicating that divalent cations have no significant role in the activity. But the necessity of divalent cations for enzyme activity has been reported for mammalian G6PD. Ni++, Co++ and Cu++ ions have been reported to inhibit G6PD (Singh et al. Reference Singh, Anand and Srivastava2012); however, Ni++ and Co++ ions increased the BmG6PD activity whereas Cu++ ions exhibited 20% inhibition. BmG6PD was robustly inhibited by Na+, K+ and Li+ ions.
The thiol groups of BmG6PD (8 Cys residues per subunit) were modified by DTNB indicating exposure of all the Cys residues in the protein. The rBmG6PD possesses 0·02 mol of SH groups per mol of monomer indicating that one of the thiol groups of G6PD is partially accessible to DTNB in the folded protein. The total number of fast-reacting SH-groups was found to be 4 per mole of rBmG6PD and thus one reactive SH group per monomer. Blocking of this group with SH-reagents inhibited enzyme activity. The different effect of pCMB and NEM indicates that some thiols susceptible to NEM modification are less accessible to pCMB and are apparently more directly involved in the catalytic activity of rBmG6PD. The modification of sulfhydryl groups by NEM probably involves conformational changes of the enzyme (Little et al. Reference Little, Sannera and Pihl1969) as indicated by changes in solubility and sensitivity to inhibition by other ligands. The recombinant G6PD of T. cruzi was unaffected by SH inhibitors as compared with the native enzyme (Igoillo-Esteve and Cazzulo, Reference Igoillo-Esteve and Cazzulo2006). DHEA, 6-AN and steroids are strong inhibitors of mammalian, T. cruzi and Plasmodium G6PD (Marks and Banks, Reference Marks and Banks1960; Cordeiro et al. Reference Cordeiro, Thiemann and Michels2009). Trypanosoma brucei G6PD was effectively inhibited by DHEA and epiandrosterone (EA) killed T. brucei bloodstream forms but these compounds showed no inhibitory effect on the recombinant Leishmania mexicana G6PD and recombinant BmG6PD. The molecular mechanism by which the steroids inhibit the G6PD of human, Trypanosoma and other lower eukaryotes including several human parasites, while they do not affect the activity of the enzyme from plants, yeasts and Leishmania, is not yet known. The specific interaction of DHEA, a non-competitive inhibitor with an enzyme–substrate ternary complex, can increase metabolic intermediates to toxic concentrations inside cells. The formation of a ternary complex is essential for the binding of non-competitive inhibitors (Gordon et al. Reference Gordon, Mackow and Levy1995). The lack of inhibition of the Leishmania G6PD and BmG6PD by DHEA and its derivatives is puzzling. It is feasible that this is due to a single but crucial substitution in the enzyme that prevents the binding of these compounds.
Since G6PD is an important enzyme that may be involved in the control of cell growth, death and redox potential of the parasites its inhibition may obstruct the cell growth and redox potential of parasites. The antifilarial drugs showed little effect on rBmG6PD activity, only suramin exhibited 75% inhibition, while CDRI compounds inhibited the BmG6PD activity at 10 μ m concentration. Among these compounds I, II and III were found to be specific inhibitors as they showed 29%, 45% and 84% inhibition, respectively. The structure of these compounds may be utilized for synthesis of novel and chemically diverse inhibitors for chemotherapy of filarial infection.
Loss of rBmG6PD activity by GdmCl/urea was found to be biphasic in nature. The guanidine at subdenaturing concentrations causes a loosening of intramolecular interaction as the secondary structure is being disrupted. GdmCl is expected to ionize into Gdm+ and Cl− ions in aqueous solution that have both ionic and chaotropic effects (Monera et al. Reference Monera, Kay and Hodges1994; Kohn et al. Reference Kohn, Kay and Hodges1995). From a structural point of view, urea and Gdm+ are very similar; however, urea is an uncharged molecule that has only chaotropic effects, whereas the guanidinium ion has a positive charge delocalized over the planar structure. At low concentrations of GdmCl, Gdm+ ions bind to negatively charged amino acid side-chains present in the protein molecule, the initial slight increase in stability due to their electrostatic interaction results in an increase of enzyme activity of rBmG6PD. The conformational stability of a protein perturbed by GdmCl/urea can be measured by both fluorescence emission and CD spectroscopy. Seven tryptophan residues are present in the BmG6PD monomer. Some of the tryptophan residues buried in the hydrophobic core of the folded protein showed an emission maximum at 340–357 nm. The GdmCl/urea-induced unfolding of enzyme occurs in 3 phases. However, significant differences in the unfolding of rBmG6PD were observed for GdmCl/urea. The first transition corresponds to the first unfolding of rBmG6PD, whereas that of the second transition corresponds to the intermediate stage and the third transition corresponds to the final phase of unfolding of the protein. The intermediate stage appears at low concentrations of GdmCl. However, at very high concentrations, GdmCl becomes a denaturant since the binding of Gdm+ ions to the protein predominates and pushes the equilibrium towards the unfolded state but in the case of urea the intermediate stage is originated for a short time. This denaturation gave some indication of 3 states of unfolding with both dimer dissociation and denaturation shifted to a higher GdmCl/urea concentration. The enzymatically inactive dimer was observed in both GdmCl/urea-induced denaturations.
In conclusion, recombinant BmG6PD showed significant differences in the kinetic properties as compared with host enzyme. rBmG6PD, a tetrameric protein, can efficiently utilize G6P as substrate and NADP+ as a cofactor. The SH-groups, 4 per mole were partially accessible in native BmG6PD. Some sulfhydryl groups were blocked by pCMB and the remaining thiol groups reacted at different rates with NEM. ATP-γ-S, ATP-β,γ-NH, ADP-β-S, Na+, K+, Li+ and Cu++ ions strongly inhibited the rBmG6PD activity. DHEA and 6-AN which are strong inhibitors of human G6PD had no effect on rBmG6PD. The triphasic pattern observed in the GdmCl/urea-treated protein indicated formation of intermediate products. The enzyme activity was significantly inhibited at low concentration of GdmCl as compared with urea. The red shift of tryptophan fluorescence to 357 nm in the presence of GdmCl/urea indicated that, upon denaturation, the tryptophan moieties are exposed. The differences observed in structure, kinetic properties and the inhibitory effect of suramin and compounds synthesized points towards the utilization of BmG6PD as a putative drug target.
ACKNOWLEDGEMENTS
We gratefully acknowledge the Council of Scientific and Industrial Research (CSIR), New Delhi, for offering a Senior Research Fellowship to Anita Verma to carry out this work. The authors are thankful to Dr T. K. Chakraborty, Director, CDRI for his invaluable support. CDRI communication no. 8392.