INTRODUCTION
Babesia caballi, a member of the phylum Apicomplexa, is a tick-borne haemoprotozoan parasite with a life-cycle that alternates between an ixodid tick host, and mammalian hosts such as the horse, in which it causes economically important diseases worldwide. In the horse, this parasite causes destruction of erythrocytes following invasion, and then induces fever, anaemia, jaundice and haemoglobulinuria. Complete prevention of B. caballi infection by drug therapy or vaccination is not currently possible (Schein, 1985; Knowles, 1988; Brüning, 1996).
B. caballi is an obligatory intraerythrocytic equine parasite. Although members of the Apicomplexa phylum infect different host and cell types, they have similar host cell invasion processes. Specifically, when an extracellular merozoite makes contact with an erythrocyte, it forms an initial reversible attachment, which leads to reorientation of the merozoite to bring the anterior apical pole in contact with the plasma membrane of the erythrocyte (Dubremetz et al. 1998; Soldati, Dubremetz and Lebrun, 2001). This in turn leads to the formation of a tight junction, through which the parasite invades the erythrocyte. Central to the invasion of host cells by Apicomplexan parasites is their employment of molecules located at the cell surface and in apical secretory organelles. These organelles are localized at the anterior end in the invasive state, at which time they are termed micronemes, rhoptries and dense granules (Dubremetz et al. 1998; Preiser et al. 2000; Blackman and Bannister, 2001).
These findings suggest that the B. caballi extracellular merozoite expresses some proteins that are important for their adhesion to and invasion of erythrocytes. To identify one of these proteins, the object of this study was to establish a mAb against a highly expressed protein of the B. caballi extracellular merozoite, to use this mAb to isolate the protein by immunoscreening of the B. caballi cDNA expression library, and to characterize the gene and its product, including its localization.
MATERIALS AND METHODS
Parasite
The United States Department of Agriculture strains of B. caballi were maintained in purified horse erythrocytes in continuous culture, as previously described (Avarazed et al. 1997; Ikadai et al. 2001).
Production of monoclonal antibody (mAb) 2H2
MAb 2H2 was produced by using a previously described method (Ikadai et al. 1999a). Briefly, 7-week-old female BALB/c mice (Clea Japan, Inc., Tokyo, Japan) were injected i.p. with 5×105 merozoites suspended in 0·1 ml of PBS emulsified with 0·1 ml of Freund's Complete Adjuvant (Difco, Michigan, USA). At 2-week intervals, 7 additional stimulations with the same amount of merozoites emulsified with 0·1 ml of Freund's Incomplete Adjuvant (Difco) were given. These mice were boosted with an inoculation of 5×105 merozoites in PBS into the caudal tail vein 2 weeks after their final immunization. The mice were sacrificed 3 days later, and their spleen cells were fused with Sp-2 mouse myeloma cells in polyethylene glycol 1500 (Roche Diagnostics, Mannheim, Germany). Hybridoma cells were selected in a GIT medium (Nihonseiyaku, Tokyo, Japan) supplemented with hypoxanthine-aminopterin-thymidine (ICN Pharmaceuticals, Ohio, USA) and maintained in GIT medium supplemented with BriClone (BioResearch, Dublin, Ireland). MAb 2H2 against a B. caballi 51 kDa protein was prepared by screening hybridoma supernatants with the indirect immunofluorescence antibody test (IFAT) and selecting a mAb 2H2 reacting with extracellular merozoite. The mAb 2H2's class and subclass was identified as IgG2a using a mouse mAb isotyping kit (Amersham Bioscience, Branchburg, NJ).
Western blotting
SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting were carried out as described previously (Ikadai et al. 1999a).
Immunoscreening of a cDNA expression library and cDNA sequencing
A B. caballi merozoite cDNA library constructed in the λ Zap II phage gene expression vector (Stratagene, La Jolla, USA) was screened with culture supernatant containing the mAb 2H2 according to the method of Ikadai et al. (1999b). Phagemids were excised from the clones and sequencing of the insert DNA of pBluescript SK (+) plasmid was performed on both strands using a Dye Terminator Cycle Sequencing Kit supplied by Applied Biosystems (Foster City, USA) with the 4 primers M13F (5′-GTAAAACGACGGCCAGT-3′), M13R (5′-GGAAACAGCTATGACCATG-3′), Bcp51F1 (5′-TACCCTCAAGTTCTTCCG-3′) and Bcp51R1 (5′-GCATGAACTTCTTGCAGTG-3′). Analysis was done with an ABI PRISM 310 DNA sequencer (Applied Biosystems) and sequencing analysis with the GENETYX-MAC Ver. 10 software (Genetyx Corp., Tokyo, Japan). The sequenced cDNA was designated as the p51 gene. Nucleotide sequence data are available in the GenBank database under Accession number AB201253.
Isolation of the p51 genomic clone
Total DNA was extracted from B. caballi by the standard method (Sambrook, Fritsch and Maniatis, 1989). B. caballi genomic DNA was amplified by PCR using one set of oligonucleotide primers, Bcp51F (5′-CATCACTTTTAACGCACCC-3′) and Bcp51R (5′-AGAGTACTCAGAGCTCAGT-3′). The primers' corresponding positions on the cDNA were 17–35 and 1460–1479, respectively, PCR was performed with Bcp51F and Bcp51R using AmpliTaq Gold DNA polymerase (Applied Biosystems) under the following conditions: 1 cycle at 95 °C for 10 min, 30 cycles at 95 °C for 1 min, 55 °C for 1 min, 72 °C for 2 min, and 1 cycle at 72 °C for 7 min. This amplified DNA was cloned into a pCR 2.1-TOPO vector using a TOPO TA Cloning Kit (Invitrogen, Carlsbad, USA). The entire ligation reaction was used to transform Escherichia coli DH5 α competent cells. Plasmid DNA from 2 positive transformants was used for DNA sequencing of the insert. The plasmid containing the gene was then isolated and subjected to DNA sequence analysis.
Northern and Southern blotting analysis
Formaldehyde-denatured total RNA (10 μg) was fractionated on a 1·2% formaldehyde–agarose gel, transferred to a nylon membrane (Hybond-N, Amersham Biosciences) and hybridized with a 32P-labelled probe derived from the p51 cDNA using the random primer DNA synthesis method in the presence of [32P]dCTP (Amersham Biosciences) (Feinberg and Vogelstein, 1983). Pre-hybridization and hybridization were performed overnight at 42 °C. Membranes were washed 3 times with 0·1× SSC (0·3 M NaCl plus 0·03 M trisodium citrate, pH 7·0) containing 0·1% sodium dodecyl sulfate (SDS) at 42 °C for 15 min. Bands hybridizing to the probe were detected by standard techniques. For Southern blotting analysis, total DNA was extracted from B. caballi by the standard method (Sambrook et al. 1989). Restriction enzyme-digested B. caballi genomic DNA was run on a 0·7% agarose gel, and the DNA was transferred onto a nylon membrane as described above. The membrane was processed and probed in the same way as for Northern blotting analysis.
Expression of p51 gene in E. coli and production of anti-GST-P51 serum
The p51 gene was amplified by PCR using one set of oligonucleotide primers, Bcp51F2 (5′-TACGCTCTCAGCCACATTT-3′) and Bcp51R (5′-AGAGTACTCAGAGCTCAGT-3′). These amplified DNAs were ligated into a cloning vector, pCR2.1-TOPO, using a TOPO TA Cloning kit (Invitrogen). The entire ligation reaction was used to transform E. coli DH5 α competent cells. The inserted p51 gene in pCR2.1-TOPO vector was subcloned into the pGEX4T plasmid (Amersham Biosciences) of the E. coli expression vector after digestion with EcoRI. The resulting plasmid pGEX-P51 was checked by sequence analysis. The pGEX-P51 was used to transform E. coli (BL21; Stratagene) by the standard technique (Sambrook et al. 1989) The recombinant protein was expressed as glutathione S-transferase (GST) fusion protein, designated GST-P51 protein.
Antiserum against the GST-P51 protein was produced in mice as anti-P51 antibody. The GST-P51 protein was purified with a MicroSpin GST Purification Module (Amersham Biosciences) after lysis of the collected bacteria by sonication in TNE (10 mM Tris–HCl [pH 7.4], 150 mM NaCl, 5 mM EDTA) containing 1% Triton-X 100. Purified GST-P51 protein (50 μg/animal) in Freund's complete adjuvant (Difco Laboratories, Detroit, USA) was intraperitoneally injected into 2 BALB/c mice aged 7 weeks. The same antigen in Freund's incomplete adjuvant (Difco Laboratories) was intraperitoneally injected into the mice on days 14, 28 and 42. Sera from immunized mice were collected 14 days after the last immunization.
IFAT
The IFAT was performed as follows. Smears of B. caballi-infected erythrocytes were prepared on slides, dried, and fixed in a 50% acetone–50% methanol solution for 5 min at −20 °C. MAb 2H2 and anti-P51 antibody were applied as first antibody on the fixed erythrocytes and incubated for 30 min at 37 °C. After 3 washes with PBS, fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG (ICN Pharmaceuticals, Aurora, USA) was applied as second antibody and incubated for 30 min at 37 °C. The slides were washed 3 times with PBS, incubated with 25 μg of propidium iodide (PI) per ml (Molecular Probes, Eugene, USA) and 50 μg of RNase A per ml for 10 min at 37 °C, and then mounted in 50% glycerol–PBS. The slides were photographed using confocal laser scanning microscopy (CLM) (TCS NT, Leica, Germany), and imaging was done using Photoshop, Version 5.0 (Adobe Systems).
Expression of p51 gene in insect cells
The p51 gene was amplified by PCR using 2 sets of oligonucleotide primers, BamBcp51, including the BamHI restriction enzyme site and ATG initiation codon (5′-ACAGTTGCAACGatgG-3′), and Bcp51Age, including the Age I restriction enzyme site (5′-TGGAGCTCAGTGTGAGGC-3′); and BamBcp51 and Bcp51stopAge, including a stop codon (5′-TGtcaGAGCTCAGTGTGAGGC-3′). One set of BamBcp51 and Bcp51Age oligonucleotide primers for PCR was used for the expressed recombinant P51 protein containing His-tag, while the second set of BamBcp51 and Bcp51stopAge was used for recombinant P51 protein without His-tag. These amplified DNAs were ligated into a cloning vector, pCR2.1-TOPO, using the TOPO TA Cloning kit (Invitrogen). The entire ligation reaction was used to transform E. coli DH5 α competent cells. The inserted p51 gene in the pCR2.1-TOPO vector was subcloned into the Autographa californica nuclear plyhedrosis virus (AcNPV) transfer vector pBlueBac4.5/V5-His plasmid (Invitrogen) after digestion with BamHI and AgeI. The resulting plasmids were designated pBlueBac 4.5/V5-p51-His and pBlueBac4.5/V5-p51-stop, respectively. Both plasmids were completely sequenced using the above-mentioned primers.
Spodoptera frugiperda (Sf9) cells were co-transfected with the recombinant transfer vectors pBlueBac4.5/V5-p51-His and pBlueBac4.5/V5-p51-stop, and linear AcNPV Bac-N-Blue DNA (Invitrogen) using the Cellfectin reagent (Invitrogen). After 6 days of incubation at 27 °C, positive blue plaques containing recominant virus were selected by a blue colour selection system. Moreover, recombinant baculovirus (Ac p51-His and Ac p51-stop) were obtained after 3 cycles of purification, respectively.
RESULTS
Cloning and sequencing of p51 cDNA clones
Screening of a cDNA expression library with mAb 2H2 against B. caballi extracellular merozoite to identify a highly expressed protein produced 5 positive clones. The nucleotide sequence of the total 1547 nucleotide cDNA was determined. Starting with methionine at position 75, a single open reading frame (ORF) of 1398 nucleotides was present. The ORF encoded a polypeptide of 465 amino acid residues with a size of 50·8 kDa. The ORF signal sequence was predicted by the algorithm of von Heijne (1986) to be the first 19 N-terminal amino acids (1MDFLAPLAFLFSVASVSFA19) of the peptide (Fig. 1A). Comparison of the deduced amino acid sequence with the GenBank database using the FASTA program showed that the B. caballi ORF encoded a protein of 465 amino acids which was 35·7% identical to the protein disulfide isomerase (PDI) of Toxoplasma gondii (GenBank Accession no. AJ3123), 34·6% identical to that of Neospora caninum (GenBank Accession no. AB178220), 32·0% identical to that of Plasmodium falciparum (GenBank Accession no. AL844507), 30·7% identical to that of Cryptosporidium parvum (Genbank Accession no. BX538350), and 26·5% identical to that of Leishmania major (GenBank Accession no. AY155573). The deduced amino acid sequence of the isolated clone exhibited an overall identity of 35·7–26·5% to proteins of PDI from several species. More specifically, this protein contained 2 distinct regions at 59–62 and 379–382 that are identical to the putative active-site sequence (Cys-X-X-Cys: CXXC) of the PDI (boldface and underlined in Fig. 1A and 1B). Moreover, the C-terminal peptides His-Thr-Glu-Leu (462HTEL465) may behave as an anchor to the endoplasmic reticulum (ER) (boldface in Fig. 1A and 1B). Based on these data, we concluded that we had isolated the B. caballi PDI gene.

Fig. 1. (A) Deduced amino acid sequence of P51 (GenBank Accession no. AB201253). Numerical positions of amino acid sequences are indicated in the right margin, and predicted signal peptide residues are underlined. PDI and thioredoxin family active sites are underlined in bold. The probable ER retention sequence (HTEL) is in bold. (B) Amino acid sequence alignments of the putative active-site sequence of the PDI protein from Toxoplasma gondii (GenBank Accession no. AJ312317), Neospora caninum (GenBank Accession no. AB178220), Plasmodium falciparum (GenBank Accession no. AL844507), Cryptosporidium parvum (GenBank Accession no. BX538350), Leishmania major (GenBank Accession no. AY155573), Saccharomyces cerevisiae (Genbank Accession no. P17967), Rattus norvegicus (GenBank Accession no. P04785) and Homo sapiens (GenBank Accession no. P07237). Asterisks and dots below the sequence denote identical and similar amino acids, respectively. (C) Schematic representation of the genomic p51 gene and Bst XI and Acc I restriction enzyme recognition sites in the p51 gene. Nucleotide sequence of the intron is shown.
Intron analysis of p51 gene
B. caballi genomic DNA was amplified by PCR using 1 set of oligonucleotide primers, Bcp51F and Bcp51R. The resulting DNA fragment was approx. 1600 nucleotides. The plasmid containing the gene was then isolated and subjected to DNA sequence analysis. The completed DNA sequence of the p51 gene was analysed and found to contain a single intron of 36 nucleotides (Fig. 1C). The sequence of the splice junctions of this intron was similar to those found in other species of protozoan parasites, such as P. falciparum PDI (Florent et al. 2000).
Characterization of P51 gene
A probe from a p51 cDNA clone was hybridized to the total RNA isolated from B. caballi merozoite by Northern blotting. The mRNA of the p51 gene is about 1·8 kb (Fig. 2A).

Fig. 2. (A) Northern blotting hybridization of the p51 gene of Babesia caballi. Tracks contained 10 μg of total RNA prepared from B. caballi-infected erythrocytes and were hybridized with the 32P-labelled p51 cDNA. The size of the markers in kilo-bases is shown at right. (B) Southern blotting hybridization of the p51 gene of B. caballi. Genomic DNA (10 μg per lane) from B. caballi-infected erythrocytes was digested with Bst XI, Acc I or Xho I, and hybridized with the 32P-labelled p51 cDNA. Size of the markers in base-pairs is shown at right.
A probe derived from the p51 cDNA clone was hybridized to B. caballi DNA fragments by Southern blotting. Genomic DNA was digested with the restriction enzymes Xho I, Acc I and Bst XI. The cDNA sequence, which did not contain a Xho I site, contained only a single Acc I site and 2 Bst XI sites (Fig. 1C). One band only was obtained on Xho I digestion, 2 on Acc I digestion, and 3 on Bst XI digestion (Fig. 2B). These results suggested that the p51 gene occurs as a single copy in the genome of B. caballi.
Expression of p51 in E. coli by pGEX-4T vector
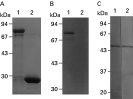
The p51 gene was ligated into the bacterial expression vector pGEX-4T, and P51 was expressed as a fusion protein with the GST protein in E. coli. The molecular masses of the GST protein and GST-P51 fusion proteins were estimated to be 27 and 78 kDa, respectively, as expected (Fig. 3A and B). Moreover, antibodies against GST-PDI fusion protein from the mice recognized only the 51 kDa native protein as mAb 2H2 (Fig. 3C). These results indicate that this 51 kDa protein did not contain the epitope of other constituent proteins in B. caballi and was unique among these proteins.

Fig. 3. Production of recombinant GST-P51 and Western blotting analysis of mAb 2H2 and anti-P51 antibody. (A) Recombinant protein samples were subjected to SDS-PAGE (10% acrylamide) and visualized by Coomassie Blue staining. Purification of recombinant GST-P51 protein (lane 1) and purified recombinant GST (lane 2). (B) Western blotting analysis of recombinant proteins, GST-P51 protein (lane 1) and GST (lane 2), probed with mAb 2H2. (C) Western blotting analysis of native Babesia caballi-infected erythrocytes with anti-P51 antibody (lane 1) and mAb 2H2 (lane 2). Molecular weights of markers in kDa are shown at left (Fig. A, B and C).
Expression of p51 in insect cells by recombinant baculovirus
Sf9 cells were infected at 5 PFU/cell with Acp51-His or Acp51-stop. After incubation for 3 days, cells infected with Acp51-His or Acp51-stop and culture media were analysed by Western blotting with anti-P51 antibody. As shown in Fig. 4, a single band of P51 protein was observed in the cell lysate with Acp51-His and with Acp51-stop, and in culture medium with Acp51-His, and the molecular mass of the P51 protein was demonstrated to be the same as that of the native B. caballi 51-kDa protein by Western blotting (Fig. 4A, lanes 1 and 2; Fig. 4B, lane 3), suggesting that the ORF in the p51 gene was the complete length. In contrast, no band was detected in the culture medium of Acp51-stop infected cells (Fig. 4B, lane 4). It remained possible that the putative ER-retention signal site (HTEL) of the recombinant protein was still functional in the ER of insect cells.

Fig. 4. Western blotting of recombinant AcP51 expressed in insect cells using anti-P51 antibody. (A) Lane 1: Babesia caballi-infected erythrocytes; lane 2: AcP51-His-infected insect cells; lane 3: AcP51-His-infected insect cell culture media. (B) Lane 1, non-infected insect cells; lane 2: non-infected insect cell culture media; lane 3: AcP51-stop-infected insect cells; lane 4: AcP51-stop-infected insect cell culture media; lane 5: B. caballi-infected erythrocytes. Molecular weights of markers in kDa are shown at left (Fig. A and B).
Cellular localization of B. caballi P51
In IFAT, the anti-P51 antibody and mAb 2H2 showed identical reactive patterns on the cold methanol-acetone-fixed preparations of B. caballi (Fig. 5). The anti-P51 antibody and mAb 2H2 reacted strongly with extracelluar B. caballi merozoites (Fig. 5A, B, G and H), and did not react in the early phase of trophozoite development (Fig. 5A and G). However, detailed observation showed that the reaction of anti-P51 antibody and mAb 2H2 were markedly irregular against pear-shaped forms, with either no reaction, or reaction to one or two brightly fluorescent pear-shaped forms (2 parasites) of B. caballi (Fig. 5D, E, F and I). Further, some trophozoites reacted to anti-P51 antibody (Fig. 5C and I). Moreover, the anti-P51 antibody reacted with the neighbourhood of the nucleus of extracellular and intracellular B. caballi merozoites (Fig. 5B and F).

Fig. 5. Confocal laser micrograph of IFAT reactive patterns. Methanol-acetone-fixed smears of Babesia caballi-infected erythrocytes were incubated with anti-P51 antibody (A–F) and mAb 2H2 (G–I). Several extracellular B. caballi merozoites (positive reaction) and earlyphase of trophozoite (negative reaction) (A, B, G and H). Trophozoites reacted to anti-P51 antibody and mAb 2H2 (C and I). Anti-P51 antibody and mAb 2H2 were markedly irregular against pear-shaped forms, with either no reaction (D and I), reaction to 1 (E) or reaction to 2 (F). Positive reactions (green) and the parasites' nuclei (red) were visualized with FITC-conjugated goat anti-mouse IgG and propidium iodide (PI), respectively.
DISCUSSION
As part of our efforts to identify new targets for chemotherapy against the parasite B. caballi, we identified the complete cDNA sequence of a highly expressed 51 kDa B. caballi extracellular merozoite protein which displays all the structural features and consensus sequences expected for a typical PDI, and corresponds to P51 of B. caballi.
Following immunoscreening with the mAb 2H2, determination of the deduced amino acid sequence of the p51 gene DNA showed homologies to the PDI of other species. The p51 gene sequence in the B. caballi genome of a single copy gene contained an intron, the splice junction of which was similar to those found in P. falciparum PDI (Florent et al. 2000). Further, the P51 protein also conserved 2 PDI Cys (CXXC) motifs and the ER retention signal site of the C-terminal. Moreover, analysis of recombinant baculovirus expression of the p51 gene in insect cells showed that its ER retention signal site was functional. In IFAT, the anti-P51 antibody reacted with the neighbourhood of the nucleus of extracellular and intracellular B. caballi merozoites. These results strongly suggest that the P51 protein is the PDI of B. caballi.
PDI is a member of the thioredoxin superfamily, which is composed of several redox proteins. It plays a key role in disulfide bond formation, isomerization and reduction within the ER, and also displays chaperone activity (Ferrari and Soling, 1999). PDI has 2 independent but non-equivalent active sites, each with 2 Cys (CXXC) that cycle between the dithiol and disulfide oxidation states, each within domains with high sequence similarity to thioredoxin (Vuori et al. 1992; Lyles and Gilbert, 1994). It is a highly abundant ER luminal protein in mammalian cells and yeast, and is essential in assisting unfolded or incorrectly folded proteins to attain their native state.
In general, PDI in eukaryotic cells have been described as having several isoforms as PDI family members. However, all of the members of the B. caballi PDI family and the mechanism by which B. caballi PDI affects the parasite in the process of invasion is also presently unknown and could not be determined in this study. Our rationale behind this search was that, from first attachment until completion of the invasion process, B. caballi secretes proteins from apical organelles into the merozoite membrane and into the environment; and that given that proteins secreted by apicomplexan parasites from micronemes, rhoptries and dense granules are generally thought to play a central role in invasion and the establishment of infection (Carruthers, 1999; Soldati et al. 2001), the adaptation of B. caballi at different stages of its development within host cells, and in the invasive process itself, may involve newly synthesized proteins or stress proteins. Therefore, one suggestion may be that B. caballi PDI plays an important role in host–parasite interactions, specifically in the optimal folding of proteins important for parasite inversion that are either secreted or expressed at the B. caballi parasite.
Support for this may come from the finding that a series of 1,4-bis (3-aminopropyl) piperazine compounds displays high activity in culture on P. falciparum, which is also one of the Apicomplexa. The homologue of the PDI of P. falciparum was isolated via a search for the plasmodial targets of this parasite by affinity chromatography using one of these compounds as a ligand (Florent et al. 2000). We therefore consider that the B. caballi PDI identified in the present study represents a new target for anti-B. caballi chemotherapy.
Moreover, mAb 2H2 recognized a 51 kDa protein produced by B. caballi. In IFAT, this protein was strongly expressed by extracellular B. caballi merozoites, and was little or not expressed in the early phase of trophozoite development. Interestingly, detailed observation showed that the expression of 51 kDa protein was markedly irregular, on fluorescence showing either no reaction, or one or two brightly fluorescent pear-shaped forms (2 parasites) of B. caballi. This result may suggest that the maturation of B. caballi merozoites after binary fission is not synchronous, and that the maturation process differs among individual merozoites.
In conclusion, the complete cDNA sequence isolated in this study encodes a protein displaying all the structural features and consensus sequences expected for a typical PDI, and corresponds to P51 of B. caballi. Further, the ER retention signal site (HTEL) of the recombinant protein retained its function in ER of insect cells. However, P51 of B. caballi was not conclusively identified as PDI in this study because we did not test the function of its protein. Thus, further studies are necessary to examine the function of this gene and the role of its protein in the process of invasion. In this regard, identification of PDI in B. caballi may facilitate the development of new inhibitors which, either independently or in conjunction with established therapies, may offer alternative treatments for B. caballi infection.
This study was supported by Grants-in-Aid for Scientific Research and Young Scientists from the Ministry of Education, Culture, Sports, Science and Technology of Japan and the Japan Society for the Promotion of Science, and the Kitasato University Research Grant for Young Researchers.