



INTRODUCTION
Genome evolution influences a parasite's pathogenicity, host–pathogen interactions, environmental constraints and invasion biology (reviewed in Garrett et al. Reference Garrett, Dendy, Frank, Rouse and Travers2006; Hoberg and Brooks, Reference Hoberg and Brooks2008; Dittmar, Reference Dittmar2009), and genome assemblies form the basis of comparative genomic analyses. These encompass not only nucleotide sequence order and gene content, but also DNA structure. Changes in genomic architecture are known to alter gene regulation affecting phenotypes (Lanctôt et al. Reference Lanctôt, Cheutin, Cremer, Cavalli and Cremer2007; Raffaele and Kamoun, Reference Raffaele and Kamoun2012), whereas structural conservation generally maintains shared features among related pathogens (Ghedin et al. Reference Ghedin, Bringaud, Peterson, Myler, Berriman, Ivens, Andersson, Bontempi, Eisen, Angiuoli, Wanless, Von Arx, Murphy, Lennard, Salzberg, Adams, White, Hall, Stuart, Fraser and El-Sayed2004; Peacock et al. Reference Peacock, Seeger, Harris, Murphy, Ruiz, Quail, Peters, Adlem, Tivey, Aslett, Kerhornou, Ivens, Fraser, Rajandream, Carver, Norbertczak, Chillingworth, Hance, Jagels, Moule, Ormond, Rutter, Squares, Whitehead, Rabbinowitsch, Arrowsmith, White, Thurston, Bringaud, Baldauf, Faulconbridge, Jeffares, Depledge, Oyola, Hilley, Brito, Tosi, Barrell, Cruz, Mottram, Smith and Berriman2007). Where repetitive elements render assembly of a given genome difficult, shared genome structure (synteny) can enhance assembly efforts (Richter et al. Reference Richter, Schuster and Huson2007; Assefa et al. Reference Assefa, Keane, Otto, Newbold and Berriman2009; Husemann and Stoye, Reference Husemann and Stoye2010). Given that closely related organisms typically maintain appreciable synteny, the genome assembly of one organism can improve the assembly of a closely related genome.
A number of interesting and important questions concerning differences among species in the parasitic genus Trichinella would benefit from high-quality genome assemblies. Nematodes belonging to the genus Trichinella are parasites that infect domestic and wild mammals that eat meat. Pork and wild game (including feral pigs, wild boars, bears, dogs, wild cats and walruses) have been implicated as sources of human infections (reviewed in Pozio, Reference Pozio2007). As such, they threaten human health and pose a concern for food safety. As most sylvatic species of Trichinella do infect humans but do not infect swine well, understanding the differences in pathogenicity and host range for species within this genus is a subject of considerable interest. In temperate North America, Trichinella spiralis and Trichinella murrelli, differ in their propensities to infect and persist in domestic swine (Kapel and Gamble, Reference Kapel and Gamble2000; Kapel et al. Reference Kapel, Webster and Gamble2005), although the bases for this difference are not understood. Trichinella spiralis has tremendous reproductive capacity in swine, and has historically been the key source of human disease via consumption of infected, undercooked pork products. Trichinella murrelli, on the other hand, has not been implicated in infections derived from pork, but exhibits a predilection for wild carnivore hosts; human infections have been acquired from bear meat (Hall et al. Reference Hall, Lindsay, Hammond, Montgomery, Wilkins, da Silva, McAuliffe, de Almeida, Bishop, Mathison, Sun, Largusa and Jones2012). Experimentally infecting swine with T. murrelli produces few muscle larvae; however, it confers some resistance to challenge with T. spiralis (D.E. Hill, personal communication, 2016). In North America, T. murrelli has been identified as the predominant Trichinella species in wild hosts (Zarlenga et al. Reference Zarlenga, Al-Yaman, Minchella and La Rosa1991). Consequently, T. murrelli threatens the health of those consuming wild game. Genetic investigations might help establish the mechanisms enforcing distinctions in their host affinities.
Genomic comparisons of T. spiralis and T. murrelli would provide the most comprehensive evaluation of the genetic differences between these two species. Until recently, a high-quality draft genome assembly was only available for T. spiralis (Mitreva et al. Reference Mitreva, Jasmer, Zarlenga, Wang, Abubucker, Martin, Taylor, Yin, Fulton, Minx, Yang, Warren, Fulton, Bhonagiri, Zhang, Hallsworth-Pepin, Clifton, McCarter, Appleton, Mardis and Wilson2011); this assembly has been used by the research community extensively (157 citations since its publication, as of this writing). In 2016, draft genomes were released for every known species of Trichinella (Korhonen et al. Reference Korhonen, Pozio, La Rosa, Chang, Koehler, Hoberg, Boag, Tan, Jex, Hofmann, Sternberg, Young and Gasser2016), providing the first opportunity for in-depth interspecific comparisons. Both published T. spiralis assemblies were constructed from short-read shotgun sequencing, resulting in average contig and scaffold lengths of 7042 and 5875 bp, respectively. Those data were derived by combining paired-end read information (Mitreva et al. Reference Mitreva, Jasmer, Zarlenga, Wang, Abubucker, Martin, Taylor, Yin, Fulton, Minx, Yang, Warren, Fulton, Bhonagiri, Zhang, Hallsworth-Pepin, Clifton, McCarter, Appleton, Mardis and Wilson2011; Korhonen et al. Reference Korhonen, Pozio, La Rosa, Chang, Koehler, Hoberg, Boag, Tan, Jex, Hofmann, Sternberg, Young and Gasser2016), fosmid end sequencing (Mitreva et al. Reference Mitreva, Jasmer, Zarlenga, Wang, Abubucker, Martin, Taylor, Yin, Fulton, Minx, Yang, Warren, Fulton, Bhonagiri, Zhang, Hallsworth-Pepin, Clifton, McCarter, Appleton, Mardis and Wilson2011), with bacterial artificial chromosome end sequencing (Mitreva et al. Reference Mitreva, Jasmer, Zarlenga, Wang, Abubucker, Martin, Taylor, Yin, Fulton, Minx, Yang, Warren, Fulton, Bhonagiri, Zhang, Hallsworth-Pepin, Clifton, McCarter, Appleton, Mardis and Wilson2011). Further consolidation of contigs into larger scaffolds or chromosomes was hindered by the approximately 20% repetitive DNA content in these genomes and by their AT-rich nature (67% adenines or thymines). Karyotypes indicate that Trichinella harbour but two autosomes and an XO sex-determination chromosome (Mutafova et al. Reference Mutafova, Dimitrova and Komandarev1982). Presently, the genomes are fragmented into thousands of sequenced components. Additional measures are needed to condense these genome assemblies towards finished genomes made up of only three chromosomes. Encouragingly, a recent analysis (Foth et al. Reference Foth, Tsai, Reid, Bancroft, Nichol, Tracey, Holroyd, Cotton, Stanley, Zarowiecki, Liu, Huckvale, Cooper, Grencis and Berriman2014) has enabled each of the 11 longest T. spiralis scaffolds to be placed into one of three chromosomes. This was accomplished by analysing single-copy genes with orthologues in the clade I whipworm Trichuris muris (Blaxter et al. Reference Blaxter, De Ley, Garey, Liu, Scheldeman, Vierstraete, Vanfleteren, Mackey, Dorris, Frisse, Vida and Thomas1998; Holterman et al. Reference Holterman, van der Wurff, van den Elsen, van Megen, Bongers, Holovachov, Bakker and Helder2006). Additional consolidation of T. spiralis contigs into chromosomes may be possible by comparing syntenic regions among contigs of closely related Trichinella species.
The prospects for success in using syntenic relationships among encapsulated species of Trichinella to aid in their assembly are favoured, given their relatively close relationships. Direct comparisons of T. spiralis and T. murrelli mitochondrial DNA revealed complete conservation of gene order, despite an average divergence of 9·5–11·8% at the nucleotide level (Webb and Rosenthal, Reference Webb and Rosenthal2011; Mohandas et al. Reference Mohandas, Pozio, La Rosa, Korhonen, Young, Koehler, Hall, Sternberg, Boag, Jex, Chang and Gasser2014). Single-copy orthologue synteny was found to be appreciable across the nuclear genome (Korhonen et al. Reference Korhonen, Pozio, La Rosa, Chang, Koehler, Hoberg, Boag, Tan, Jex, Hofmann, Sternberg, Young and Gasser2016). The two species are thought to have shared a common ancestor between 7 and 10 million years (MY) ago, based on comparisons of nuclear and mitochondrial gene sequences (Zarlenga et al. Reference Zarlenga, Rosenthal, Rosa, Pozio and Hoberg2006; Korhonen et al. Reference Korhonen, Pozio, La Rosa, Chang, Koehler, Hoberg, Boag, Tan, Jex, Hofmann, Sternberg, Young and Gasser2016). Furthermore, a higher degree of synteny has been reported among encapsulated trichinellids (including T. spiralis and T. murrelli) than non-encapsulated species, with a syntenic correlation of 0·118 for genes in the nuclear genomes of T. spiralis and T. murrelli (Korhonen et al. Reference Korhonen, Pozio, La Rosa, Chang, Koehler, Hoberg, Boag, Tan, Jex, Hofmann, Sternberg, Young and Gasser2016). Therefore, in order to improve Trichinella genome assemblies, we used third-generation, long-read technology to sequence through highly repetitive regions of the genome. We sequenced the genome of the North American sylvatic species, T. murrelli, using long-read technology and generated a de novo assembly which was polished using high-quality base calls derived from Illumina paired-end sequencing. This sequence was exploited to improve the assemblies of both T. murrelli and T. spiralis. Using syntenic information between the two assemblies, T. murrelli contigs were placed into scaffolds representing putative chromosomes. Additionally, T. spiralis contigs were added to chromosome groupings based on synteny with the T. murrelli assembly. Finally, certain regions of T. spiralis scaffolds were reoriented based on putative scaffolding errors revealed by comparisons with longer T. murrelli contigs achieved through long-read sequencing. As a result, we achieved a 10-fold improvement in overall genome assemblies of both T. murrelli and T. spiralis.
MATERIALS AND METHODS
Genome sequencing and assembly
Trichinella murrelli isolate ISS35 was used as the source of genomic DNA for sequencing. This isolate was collected from a black bear (Ursus americanus) in North America in 1982, and had been passed through mice approximately twice per year since its isolation. In 2011, Swiss-Webster mice were infected with T. murrelli ISS35, and worms were collected from eviscerated mouse carcasses using standard digestion procedures. In order to minimize host DNA contamination, collected worms were pre-treated with DNAse I for 10 min and washed extensively in tap water prior to parasite DNA extraction. Following digestion with proteinase K in 10 mM Tris pH 8.5 with 0.5% sodium dodecyl sulfate (SDS), nucleic acids were collected by phenol–chloroform extraction and ethanol precipitation. The sample was then treated with a cocktail of RNAse A and RNAse T1. The final genomic DNA preparation was checked for purity using gel electrophoresis and spectrophotometric analysis by NanoDrop. Genomic DNA was quantified using the Stratagene Quantifluor system.
Trichinella murrelli genomic DNA was sequenced using Pacific Biosciences (PacBio) (Menlo Park, CA) long-read technology at the University of Maryland School of Medicine's Institute for Genome Sciences. One PacBio Ultra Long Insert Library was generated. This library was loaded onto four PacBio RS II P6-C4 SMRT-cells in order to generate long-read sequences. Long-read sequencing generated 334 806 reads averaging 9756 bp in length (s.d. = 6637) for a theoretical 50X coverage of the expected 65 Mbp genome.
Long reads were assembled using SMRT Analysis v. 2.3.0, assuming a total assembly size of 65 Mbp (based on the estimated size of the 2011 T. spiralis genome assembly). Base calls in the long-read assemblies were improved using Illumina paired-end short reads of 300 bp, obtained using the v3 run capabilities of an Illumina MiSeq, which provided a theoretical 150X coverage prior to quality control of reads. Each Illumina read was trimmed such that no more than two nucleotides within each read had a Q score of <30 (0·1% chance of erroneous base call). Illumina paired-end reads were then aligned to the long-read contigs using the Geneious assembler (Geneious v. 8.0.6, http://geneious.com (Kearse et al. Reference Kearse, Moir, Wilson, Stones-Havas, Cheung, Sturrock, Buxton, Cooper, Markowitz, Duran, Thierer, Ashton, Meintjes and Drummond2012)) with medium–low sensitivity and five iterations, resulting in an average of 80X coverage for each base in the SMRT Analysis assembly. To assign nucleotides for the final assembly, consensus base calls were derived from Illumina reads where SMRT assembly contig coverage was greater than 10X. In order to call a base as a heterozygote, minority reads had to account for >10% of all Illumina base calls at that locus. For those sites with less than 10X coverage, the original SMRT Analysis base call was retained.
Repetitive DNA was identified using a combination of ab initio repeat finding using RepeatModeler v. 1.0.8 (Smit and Hubley, Reference Smit and Hubley2008) and a library-based approach in RepeatMasker v. 4.0.5 (Smit et al. Reference Smit, Hubley and Green2013). Within RepeatModeler, ab initio analysis was conducted in RepeatScout v. 1.0.5 and RECON v. 1.0.8 to produce a library of repeat elements. RepeatMasker was then used to identify each individual repeat in the assembly contigs and classify it as either a simple repeat or as an interspersed repetitive element. RepeatMasker was also used independently, specifying T. spiralis repeat elements from Repbase, including the Ginger2 DNA transposon, the R2 family of non-LTR retrotransposons, the Utopia family of non-LTR retrotransposons, and the TSRP-1 repetitive sequence. Finally, Phobos was used to search for degenerate simple repeats fewer than 5 nt in length, allowing for 20% divergence from the core repeat pattern. Subsequent analyses regarding alignment of T. murrelli contigs with T. spiralis scaffolds were conducted using the masked DNA file output from RepeatMasker so as to minimize errors and ambiguities that typically ensue when repeats are included.
The new long-read assembly was compared with existing Trichinella genome assemblies using QUAST v.4.0 (Gurevich et al. Reference Gurevich, Saveliev, Vyahhi and Tesler2013) for length metrics and CEGMA v.2.4.010312 (Parra et al. Reference Parra, Bradnam and Korf2007) and BUSCO v.2.0 (Simão et al. Reference Simão, Waterhouse, Ioannidis, Kriventseva and Zdobnov2015) in order to examine completeness of coding regions in the genome. Default QUAST settings were used to determine the shortest contig or scaffold that was required to encompass 50% (N50) or 90% (N90) of the total length of all contigs or scaffolds for each assembly in the comparison. CEGMA analysis was conducted in the Cyverse.org Discovery Environment (Merchant et al. Reference Merchant, Lyons, Goff, Vaughn, Ware, Micklos and Antin2016) using the 248 most highly conserved core eukaryotic gene set (CEGs). The new long-read T. murrelli assembly, the 2011 T. spiralis assembly and the 2016 T. murrelli short-read assembly were analyzed for complete, partial and missing CEGs. BUSCO was used to examine single-copy orthologues (BUSCOs) associated specifically with nematodes for both the novel long-read T. murrelli assembly and the 2016 T. murrelli short-read assembly. The BUSCO.py program was run with the ‘nematoda_odb9’ lineage in genome mode with species set to ‘Caenorhabditis’, and complete, duplicated and fragmented BUSCOs were reported.
Synteny and chromosomal scaffolding
ProgressiveMAUVE v. 20150226 (Darling et al. Reference Darling, Mau and Perna2010) was used to derive an initial overview of synteny between the T. murrelli corrected contigs and 2011 T. spiralis scaffolds. Using 2011 T. spiralis scaffolds as reference sequences, T. murrelli contigs were reordered using the Move Contig feature in progressiveMAUVE to achieve optimal alignment. Alignments of long collinear blocks (LCBs) were visualized in the MAUVE viewer, relying on LCBs with a weight >10 000 to assess synteny. LCBs with lesser weight were reserved for MUMmer analysis outlined below.
In order to maximize the organization of both assemblies, 2011 T. spiralis scaffolds were divided into putative chromosomes according to single-copy orthologue analyses (Foth et al. Reference Foth, Tsai, Reid, Bancroft, Nichol, Tracey, Holroyd, Cotton, Stanley, Zarowiecki, Liu, Huckvale, Cooper, Grencis and Berriman2014). Chromosome 1 comprised scaffolds GL622784, GL622787, GL622790, GL623393 and GL624340 of the 2011 T. spiralis assembly. Chromosome 2 encompassed scaffolds GL622785, GL622788 and GL623868, while scaffolds GL622791, GL622792 and GL622789 were designated as Chromosome X.
MUMmer v. 3.23 was used to examine local areas of synteny among T. murrelli masked contigs and T. spiralis putative chromosomes. Using the NUCmer application within MUMmer, maximal unique matches (MUMs) were identified between T. murrelli contigs >7000 bp (60% of the total length of all T. murrelli contigs) and each putative T. spiralis chromosome. Each group of T. murrelli contigs was then placed in optimal alignments relative to T. spiralis scaffolds using the move contig feature in MAUVE. These optimized T. murrelli putative chromosomes were concatenated into scaffolds using 100 indeterminate bases (‘N’) to signify gaps between contigs that occur within any T. spiralis scaffold, and 1000 bases between contig ends that occur between T. spiralis scaffolds signifying less confidence in the ordering of the final placement of T. murrelli contigs in the chromosomal scaffolds. Subsequently, 2011 T. spiralis scaffolds were broken at assembly gaps for reassembly based on synteny with the new T. murrelli chromosomes. A custom PerlScript was written to identify stretches of N's that were equal to 10 nucleotides or longer, break the scaffold at each such instance, and place each sub-sequence corresponding to the sequence between adjacent assembly gaps into a new FASTA file, retaining the N's present in the original scaffold. This process was intended to retain as much information as possible from the original scaffolding efforts of Mitreva et al. (Reference Mitreva, Jasmer, Zarlenga, Wang, Abubucker, Martin, Taylor, Yin, Fulton, Minx, Yang, Warren, Fulton, Bhonagiri, Zhang, Hallsworth-Pepin, Clifton, McCarter, Appleton, Mardis and Wilson2011). The resulting sequences are heretofore termed ‘T. spiralis contigs’. After breaking the T. spiralis scaffolds into their component parts, these T. spiralis contigs were matched to putative T. murrelli chromosome sequences using MUMmer. It was expected that this reverse matching would correctly identify not only sequences from the original 11 long T. spiralis scaffolds, but contigs that belong to each chromosomal group that were not hypothesized by Foth et al. (Reference Foth, Tsai, Reid, Bancroft, Nichol, Tracey, Holroyd, Cotton, Stanley, Zarowiecki, Liu, Huckvale, Cooper, Grencis and Berriman2014), thus lengthening chromosomal sequences of the T. spiralis assembly.
All statistical analyses were conducted in R. The distribution of contig/scaffold lengths was determined using the density function. Median coverage of T. murrelli contigs was determined using Rsamtools, extracting read counts from BAM files of short-reads mapped to T. murrelli long-read contigs. Output of median coverage was graphed in Microsoft Excel.
RESULTS
Subjecting an isolate of T. murrelli to long-read sequencing using the Pacific Biosystems technology generated 334 806 individual reads averaging 19·6 kbp in length; the longest read extended 51 413 bp. Approximately 3% of the reads corresponded to adapter dimers and inserts <100 bp. Following filtering, nearly 224 000 reads were deemed appropriate for use in de novo assembly. These reads averaged 13 786 bp, with an N50 of 19 125, and an average read score (probability of being correct) of 0·85.
After filtering for quality, long reads were assembled into a high-quality draft genome with a total size of 63·2 Mbp (Table 1). Over half of the total assembly's length was derived from just 26 contigs, each of which is longer than 571 000 bp (N50). The N90 is 27 652 bp, and the largest contig is over 3.6 Mbp. Short-read, paired-end sequences were used to verify and correct small errors in the assembled contigs. Paired-end reads of 300 bp were aligned to T. murrelli assemblies, providing 80X coverage on average; consensus sequence of the final contigs was determined by those short-read base calls comprising >10% of the coverage. Nearly all of the corrections involved the insertion or deletion of a single nucleotide, following a run of that same nucleotide. Heterozygous bases accounted for 0·5% of all nucleotides or 1 heterozygous base every 200 bp. Attempts at improving the assembly via scaffolding using SSPACE did not further reduce the number of contigs.
Table 1. Trichinella genome assembly sources and statistics

a As reported by Korhonen et al. (2016).
Based on Recon and RepeatScout ab initio libraries, RepeatMasker identified 13 843 interspersed repeat elements within the final contigs, accounting for 15 795 525 bp (25%) of the T. murrelli genome. Additionally, RepeatMasker analysis indicated that 2·7% of the genome could be categorized as short simple repeats. Phobos analysis, allowing for 20% degeneracy in any repeat motif, found 3 355 004 bases (5·3%) within tandem repeats with a 5 nucleotide repeat motif or less (nearly twice as many as detected by RepeatMasker), bringing the total amount of repetitive DNA to 30·3%. Repetitive elements were sub-divided into seven categories. There were 21 SINEs, 929 LINEs, 2856 LTR, 3281 non-LTR DNA elements, 6596 low complexity sequences, 160 satellite elements and 36823 simple repeats. These were spread across all T. murrelli contigs, but were at highest concentration in contigs <100 000 bp in length. In order to determine where the repeat sequences were concentrated, we plotted repeat content as a function of contig length. Repeats comprised a smaller proportion of contigs larger than 100 000 bp (Student's t-test, P < 0·0001); the variance in repeat composition, which ranged from 8 to 58% in contigs greater than 100 000 bp, was reduced when compared with smaller contigs (Fig. 1).

Fig. 1. Proportion of each long-read T. murrelli contig that is occupied by repetitive DNA elements. Those contigs greater than 100 000 bp in length (black diamonds) have much more consistent representation of repetitive DNA than smaller fragments (grey circles). The general lower proportion of repetitive sequences may contribute to the ease with which these portions of the genome are assembled. This would lend some greater confidence to these longer contigs when trying to understand reorganization within the genome relative to other closely related genomes.
A survey of the core eukaryotic genome (CEGs) was conducted to assess the completeness of the protein coding portions of the assemblies. CEGMA analysis indicated that the 94·0% of CEGs were complete in the new T. murrelli long-read assembly, a minor improvement over the 93·6% completeness of the 2011 T. spiralis assembly. Using other search parameters, the 2016 short-read assembly of T. murrelli (Korhonen et al. Reference Korhonen, Pozio, La Rosa, Chang, Koehler, Hoberg, Boag, Tan, Jex, Hofmann, Sternberg, Young and Gasser2016) had been reported to have 96·0% of CEGs complete. However, subjecting that assembly to search parameters identical to those used above, we found the 2016 T. murrelli short-read assembly to have 92·7% of CEGs complete. Similar improvements were found in the new assembly when employing BUSCO to determine the number of nematode-specific single-copy orthologues recovered in each. BUSCO found that 88·8% of nematode single-copy orthologues were complete in the novel long-read assembly whereas the 2016 short-read assembly revealed only 78·2% of single-copy orthologues that were complete (Table 1). Therefore, each assembly was judged successful in assembling areas of the genome carrying coding information, with a 10% improvement in recovering nematode-specific single-copy orthologues in the long-read assembly.
Comparison of T. murrelli long-read genome assembly with 2011 T. spiralis assembly and 2016 short-read T. murrelli assembly
The T. murrelli assembly presented here represents approximately a 10-fold improvement over the published 2011 T. spiralis genome assembly at the contig level. The 2011 assembly of T. spiralis in GenBank (Accession # ABIR02·1, accessed 12/9/2015), which was assembled via whole genome shotgun sequencing and hierarchical mapping based on fosmid and BAC end-sequencing, contained 6859 scaffolds comprising 9261 contigs. This T. spiralis assembly, with a total length of 63 521 838 bp, had an N50 of 76 808 and an average length of 9261 bp/scaffold. Of those scaffolds, 3008 were smaller than 1000 bp. Similarly, the recently published short-read T. murrelli assembly comprised 5255 scaffolds with an N50 of 106 482 bp with 3913 scaffolds <1000 bp in length. Notably, the short-read T. murrelli assembly totalled just over 49 Mbp, suggesting that over 14 Mbp were not accounted for in comparison with the 2011 T. spiralis assembly or the long-read T. murrelli assembly presented here. By contrast, our long-read T. murrelli assembly is condensed into nearly 15-fold fewer total contigs (Fig. 2A), and eight times as many of its contigs are larger than 1000 bp (see distribution of scaffold/contig lengths for each assembly in Fig. 2B). Our long-read T. murrelli N50 is 7·5 times the size of the 2011 T. spiralis N50 and five times longer than the recently published short-read assembly of T. murrelli, achieving an average contig length 13·9 times larger for T. murrelli than for 2011 T. spiralis scaffolds. Almost 8% of positions had been designated as N (indeterminate sequences) within the scaffolds of the 2011 T. spiralis assembly, but fewer than 0·1% of the positions in our long-read T. murrelli assembly are ambiguous in this way. The recently published short-read T. murrelli assembly had small numbers of indeterminate bases (0·6%) but, as noted previously, had 22% less total sequence than the assembly provided here.

Fig. 2. Comparative summary of Trichinella genome assemblies. (A) Cumulative length of contigs or scaffolds when arranged from longest (contig index 1) to shortest for each assembly as implemented in Quast. The T. murrelli long-read assembly reaches complete length in many fewer contigs than either the 2011 T. spiralis or 2016 T. murrelli short-read assembly. (B) For the 2011 T. spiralis and 2016 T. murrelli short-read assemblies, 91·6% and 83·3% of the contigs are smaller than 3000 bp, respectively; only 2·2% of contigs in the long-read T. murrelli assembly are that small. The 63% of all T. murrelli long-read contigs fall between 20 000 and 50 000 bp. An additional 10% of long-read T. murrelli contigs fall between 200 000 and 500 000 bp. The long-read T. murrelli assembly has substantially improved N50 (571 829 bp) (Table 1), despite the fact that the longest T. spiralis scaffold (GL622787 = 12·1 Mbp) is over three times the length of the longest T. murrelli contig (unitig 1112 = 3·6 Mbp).
Alignment of the T. murrelli assembly presented here and the 2011 T. spiralis assembly showed remarkable overall synteny. After reordering the T. murrelli contigs in MAUVE in order to maximize alignment of T. murrelli contigs with T. spiralis scaffolds, 343 LCBs weighted >10 000 were observed (Fig. 3). At the broadest level, approximately 51·5 Mbp (81·7%) of the assemblies were collinear (ignoring gaps between collinear blocks). Similarly, MUMmer alignment of the 2011 T. spiralis assembly and 2016 T. murrelli long-read contigs resulted in a total of 51·0 Mbp of aligned sequence, nearly identical to that assessed by alignments in MAUVE. Identity among local MUMmer alignments ranged from 55 to 100% with an overall 92·4% identity across all alignments. Maximal unique matches ranged in size from 67 to 70 998 bp in the masked alignment. For those MUMs larger than 10 000 bp (signifying strong confidence in the correct assignment of orthologous regions in the two genomes), the overall identity was 92·6% and accounted for 48·5% of the total length of aligned sequences. There remained 472 T. murrelli contigs that could not be aligned with any confidence; these averaged 20 902 bp in length and were enriched for repetitive sequences. Similarly, 6841 T. spiralis scaffolds could not be aligned; these averaged 1561 bp in length. A comparison of the sizes of contigs/scaffolds that could be aligned with those that could not be is shown in Fig. 4. Those contigs and scaffolds that could not be aligned confidently were not included in subsequent chromosomal organization.

Fig. 3. ProgressiveMAUVE alignment of 2011 T. spiralis and T. murrelli long-read assemblies using the ‘Move Contig’ feature to optimize placement of T. murrelli contigs relative to the 2011 T. spiralis scaffolds. Approximately 51·5 Mbp were located within LCBs, accounting for 82% of the entire assemblies. Arrows indicate the location of local syntenic rearrangements wherein collinear blocks have the same orientation, but have local rearrangements such that each block is precisely reversed in the T. murrelli assembly with respect to the T. spiralis assembly. Note that each rearrangement occurs within an intact T. murrelli contig, denoted by the vertical red lines of the lower assembly. There are 25 different regions of the alignment that follow this pattern.

Fig. 4. The distribution of T. spiralis (Tsp) scaffold and T. murrelli (Tm) contig lengths divided into those that could or could not be confidently aligned in initial MAUVE and MUMmer alignments. For each species, those sequences which could not be aligned were significantly smaller than those that aligned with confidence (Student's t-test, P<0·01 for both comparisons).
The chromosome hypothesis
Foth et al. (Reference Foth, Tsai, Reid, Bancroft, Nichol, Tracey, Holroyd, Cotton, Stanley, Zarowiecki, Liu, Huckvale, Cooper, Grencis and Berriman2014) hypothesized how the largest scaffolds in T. spiralis should be grouped into chromosomes, using single-copy orthologues mapped to the whipworm, T. muris. In order to assess whether our data supported the organization of contigs into chromosomes, we examined median short-read coverage of T. murrelli contigs, expecting coverage of the sex chromosome to be 3/4 the coverage of autosomal sequences (as would be true for an XO sex-determining system in the case where male and female DNA were equally represented in the sequenced template). The coverage of 57 long-read T. murrelli contigs ranged between 94 and 111 reads/site, whereas the coverage of another 52 contigs ranged between 72 and 87 reads/site; we supposed that these distinct coverage groupings might correspond, respectively, to the autosomes and to the X chromosome (Fig. 5). Another 545 contigs, mapped fewer than 70 reads/site, were ignored for the purposes of this analysis. Confirming this supposition, all of the contigs so classified as autosomal also matched sequences aligning with autosomal sequences of T. spiralis in the MUMmer analyses. Of the contigs classified as belonging to the X chromosome, 83% matched sequences designated as residing on the X chromosome of T. spiralis by Foth et al. Reference Foth, Tsai, Reid, Bancroft, Nichol, Tracey, Holroyd, Cotton, Stanley, Zarowiecki, Liu, Huckvale, Cooper, Grencis and Berriman2014. The nine exceptions (contigs that matched sex chromosome coverage ratios, but had been judged previously to be autosomal by single-copy orthologue content) merit further analysis to assess their chromosomal assignment. Interestingly, most contigs whose coverage did not match chromosomal expectations are shorter than 100 000 bp and harbour much repetitive DNA, which may compromise attempts at ascertaining their chromosomal location. The evidence from two independent lines justified organizing most of these data into one of three chromosomes.

Fig. 5. Median short-read coverage of T. murrelli contigs supports hypothesized division of contigs into autosomal and sex chromosome groups. Trichinellids have an XO sexual determination system; if male and female genomes contributed equally to the template used in chromosome sequencing, then reads corresponding to the X chromosome should be 3/4 as frequent as those mapping to either of the two autosomes. (A) The actual distribution of median read coverage for T. murrelli contigs by short-read Illumina sequences closely matches this expectation based on the 109 contigs with the highest median coverage, in that read densities either cluster at ~105 reads/base (blue squares) or at ~75 reads/base (purple triangles). Those 544 contigs with median coverage <67 reads/bp (not shown) were considered outside of the normal range required for this analysis. (B) Most contigs (92·7%) assigned as putative autosomal or X chromosome sequences matched their chromosomal assignment by homology to T. spiralis scaffolds. However, five contigs matching T. spiralis chromosome 1 sequences (yellow squares) and three contigs matching chromosome 2 sequences (red circles) disagreed with homology assignments and require further study for confident placement on chromosomes.
Employing the chromosomal framework of Foth et al. Reference Foth, Tsai, Reid, Bancroft, Nichol, Tracey, Holroyd, Cotton, Stanley, Zarowiecki, Liu, Huckvale, Cooper, Grencis and Berriman2014, we assigned scaffolds of T. spiralis into their respective chromosomes. We placed homologous T. murrelli long-read contigs into corresponding chromosomal files. We then optimized the order of long-read T. murrelli contigs using the ‘move contig’ process of MAUVE, relying on the original T. spiralis scaffolds as a baseline for organization; the resulting T. murrelli and T. spiralis hypothetical chromosomes are depicted in the upper portion of each panel in Fig. 6 and denoted as ‘Before’, signifying that T. spiralis sequences had not yet been optimized relative to long-read T. murrelli contigs. While some local inconsistencies persisted among the T. spiralis scaffolds and T. murrelli contigs, the general agreement between the two assemblies indicates that scaffolding of T. murrelli contigs based on T. spiralis scaffolds was tenable. This resulted in three T. murrelli chromosomal scaffolds with a total size of 52·7 Mbp, and increased the scaffold N50 to 17·1 Mbp. These results represent our current best evidence for chromosome organization for T. murrelli.
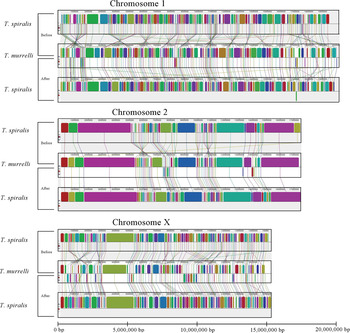
Fig. 6. Depiction of putative chromosomes before and after realignment. In the upper portion of each chromosome panel, T. murrelli contigs from each putative chromosome have been moved to optimally match 2011 T. spiralis scaffolds predicted to correspond to each chromosome. In the lower part of each chromosomal depiction, T. spiralis scaffolds were broken at assembly gaps into component contigs, then contigs were reassembled into putative chromosomes based on MUMmer matches to the newly established T. murrelli chromosomes. The decrease in crossing LCB connecting lines is an indication of better syntenic arrangements along putative chromosomes. The final chromosomal scaffolds provide our best estimate of genome sequences for both species based on positional evidence derived from long-read uninterrupted T. murrelli contigs and scaffolding information present in the 2011 T. spiralis assembly.
Certain segments of the MAUVE alignments between T. murrelli and the 2011 T. spiralis genome revealed a highly consistent criss-cross pattern (highlighted with arrows in Fig. 3) that might be mistaken for large genomic inversions. A simple inversion would have reversed the orientation of those LCBs within such segments, but close inspection of these regions showed them to retain their orientation, despite the reversal of LCB order (Fig. 7). This incongruous pattern always occurred within a single T. murrelli contig but among T. spiralis contigs that had been connected by ‘assembly gaps’ in the 2011 T. spiralis assembly scaffolding process. If biological in origin, this result would require rampant, local rearrangements in the order of LCBs. Misassembly across assembly gaps in 2011 T. spiralis scaffolds would offer a simpler explanation for this pattern.

Fig. 7. Graphic showing the difference between the observed LCB alignment pattern and a classic chromosomal inversion. Each coloured block corresponds to an LCB determined by MAUVE. The direction of the arrows indicate the direction of the aligned sequences. In a classical inversion, the segment ABCDEF would be reversed (FEDCBA) and the arrows would point in the opposite direction of the reference sequence. What was observed was that the order of the LCBs was reversed (FEDCBA), but oriented as in the reference sequence, indicating that the LCBs were reordered, but not reversed. Such a rearrangement would have required at least six separate translocations of individual LCBs while retaining regional cohesion within the chromosome. This seems unlikely in closely related species.
We therefore reoriented LCB's in the 2011 assembly of T. spiralis to conform to the LCB orders indicated by long-read T. murrelli contigs. For each set of scaffolds belonging to a putative chromosome (1, 2 or X), we deconstructed T. spiralis scaffolds into their component contigs by breaking them at assembly gaps of 10 bp or longer. We then aligned these contigs to T. murrelli chromosome scaffolds, and substantially greater agreement resulted. Finally, we reinserted assembly gaps between T. spiralis contigs so as to match the total length of the original scaffolds, preserving spacing information gleaned from the 2011 assembly. As a result, we were able to condense the assembly of T. spiralis into fewer component pieces with improved confidence in ordering within scaffolds (Fig. 6, ‘After’).
Pairs of assembled genomes often require transpositions and inversions in order to be fully reconciled; fewer such rearrangements are required to harmonize highly syntenic assemblies. The procedures described above markedly reduced the number of such rearrangements between genome assemblies of two encapsulated members of the genus Trichinella. When initially compared, our assembly of T. murrelli required 35 inversions and 197 transpositions of LCBs in order to be reconciled with the 2011 assembly of T. spiralis (an inversion to transposition rate of 1:5·6). Of these, 24 LCB inversions and 155 LCB transpositions occurred on autosomes, whereas 11 inversions and 42 transpositions on putative sex chromosomes. After identifying and correcting for putative local inconsistencies in the 2011 assembly of T. spiralis, the total number of inversions and transpositions dropped to 7 and 14, respectively. All seven of these remaining inversions occurred on autosomal sequences, as did 11 of the 14 transpositions. This resulted in an inversion:transposition ratio of 1:1·6 for autosomes and 0:3 for the X chromosome. Assuming two chromosome breakages for each inversion and three for each translocation as well as 7–10 MY of divergence time, this translates to 0·15 to 0·21 chromosome breakages/Mbp/MY.
DISCUSSION
We sequenced the genome of the sylvatic Trichinella species, T. murrelli, using third-generation, single-molecule, long-read technology. The final assembly condensed to 653 contigs; half of the assembled genome occurred in contigs longer than 571 031 bp (N50). This represents an approximately 10-fold improvement over the 2011 assembly of the T. spiralis genome in terms of length and completeness. The genome assemblies were generally in agreement with 51·5 Mbp of contiguous sequence. Trichinella murrelli long-read contigs spanned many gaps in the 2011 T. spiralis scaffolds, providing great empirical support for the T. murrelli contigs; we therefore used these to reorder the 2011 T. spiralis assembly, resulting in an improved 2016 T. spiralis assembly. Furthermore, we were able to use two lines of evidence to situate sequences of T. murrelli and T. spiralis into their two autosomes and one X chromosome.
The genomes of closely related species often retain syntenic organization. While translocations and inversions occur, chromosomal gene content is often conserved, particularly among nematodes (Guiliano et al. Reference Guiliano, Hall, Jones, Clark, Corton, Barrell and Blaxter2002; Hillier et al. Reference Hillier, Miller, Baird, Chinwalla, Fulton, Koboldt and Waterston2007; Simakov et al. Reference Simakov, Marletaz, Cho, Edsinger-Gonzales, Havlak, Hellsten, Kuo, Larsson, Lv, Arendt, Savage, Osoegawa, de Jong, Grimwood, Chapman, Shapiro, Aerts, Otillar, Terry, Boore, Grigoriev, Lindberg, Seaver, Weisblat, Putnam and Rokhsar2013). Here, two assemblies from species of Trichinella were used to improve each other. The 2011 T. spiralis scaffolds provided valuable insight into long-range association of contigs, derived from BAC end-sequencing. Furthermore, information from single-copy orthologues allowed scaffolds of T. spiralis, and then T. murrelli, to be assigned to chromosomes. Conversely, the long de novo contigs comprising the T. murrelli assembly identified the need to reorder LCBs in T. spiralis. These data justified the conclusion that the conspicuous crossing pattern in macrosyntenic alignments had not resulted from rampant inversions, but rather from frequent local misassembly of contigs in the 2011 T. spiralis assembly, separated by sequencing gaps. We cannot explain why these blocks appeared in reverse order, but we are confident that the source of conflict was computational, not biological. Thus, the final assembly of T. murrelli using long reads allowed assembly limitations imposed by short reads in highly repetitive genomic regions to be overcome. While the ordering of scaffolds within chromosomes still requires refinement, the sequences resulting from this effort provide a basis for future investigations of inherited linked genes.
Assembly of genomes is most difficult when derived from short reads and when highly repetitive elements force assembly ambiguities; this limitation was largely overcome by obtaining sequence reads far longer repeat intervals. Doing so substantially increased contig length and decreased their number. We achieved an approximately 10-fold improvement over the T. spiralis assembly published in 2011 as well as the 2016 short-read T. murrelli assembly. We placed the most confidence in contigs >100 000 bp; these exhibited more consistent read coverage and repetitive sequence composition. These longer contigs accounted for 82·2% of the entire assembly length. The longest T. murrelli contig spanned 116 gaps in the 2011 T. spiralis assembly; these gaps totalled 369 885 bp of the 3·6 Mbp total. In addition, long-read sequencing reduced indeterminate bases 80-fold (from 8 to <0·1%); the longer reads confirmed the scaffolding of some gaps, but revealed many errors in the scaffolding process used previously. By assuming synteny, we were able to leverage these longer contigs in T. murrelli to improve scaffolding in the assembly of T. spiralis.
Nevertheless, the 2011 T. spiralis scaffolds, achieved through hierarchical scaffolding, contain useful proximity information which we used to orient the T. murrelli contigs into putative chromosomes, enabling our new scaffolding hypothesis for T. spiralis based on alignments between T. murrelli contigs and ungapped T. spiralis contigs. While further work is required to place the contigs that did not match cleanly with chromosomal sequences, long-read sequencing substantially improved the quality and confidence of both genome assemblies.
We were able to situate our scaffolds into each of two autosomes and one sex-determining X chromosome based on two lines of evidence, single-copy orthologues as per Foth et al. Reference Foth, Tsai, Reid, Bancroft, Nichol, Tracey, Holroyd, Cotton, Stanley, Zarowiecki, Liu, Huckvale, Cooper, Grencis and Berriman2014, and median depth of short-read coverage of our long-read assembly. MUMmer was then used to determine which T. murrelli contigs aligned with a given T. spiralis putative chromosome, and allowed for informed scaffolding into new putative T. murrelli chromosomes. Illumina short reads were aligned to the T. murrelli contigs and median depth of coverage per base pair was used to test whether putative X chromosome coverage differed from autosomal coverage. Unless only females are sequenced, X chromosomes should be covered by fewer reads than autosomes. The extent of this expected deficit depends on the sex ratio (3/4 for a population comprised of equal numbers of male and female genomes); but the ratio of X:autosome would be 5:6 if, as has been reported, females generally outnumber males by nearly 2:1 (Rappaport, Reference Rappaport1943; Gursch, Reference Gursch1949; Boyd and Huston, Reference Boyd and Huston1954; Boyd and Huston Reference Boyd and Huston1954, Gursch, Reference Gursch1949; Rappaport, Reference Rappaport1943). The actual ratio approximated 3:4 based on median coverage, in line with expectations. While nine contigs were classified as having X chromosome coverage but linked to autosome sequence based on gene content, median short-read coverage of contigs provided a reasonable method for preliminary separation of sex chromosome contigs from autosomes.
Long-read assembly of T. murrelli substantially refined the estimate of repeat content in Trichinella genomes. Our estimate of repetitive DNA content was 12% higher than those made by both Mitreva et al. Reference Mitreva, Jasmer, Zarlenga, Wang, Abubucker, Martin, Taylor, Yin, Fulton, Minx, Yang, Warren, Fulton, Bhonagiri, Zhang, Hallsworth-Pepin, Clifton, McCarter, Appleton, Mardis and Wilson2011 and Korhonen et al. Reference Korhonen, Pozio, La Rosa, Chang, Koehler, Hoberg, Boag, Tan, Jex, Hofmann, Sternberg, Young and Gasser2016. Conflict in these estimates may derive either from the large proportion of indeterminate sequences present as assembly gaps in the 2011 T. spiralis assembly, or from the 14 Mbp of sequence not documented in the 2016 T. murrelli short-read assembly. However, because of the long sequence reads presented herein, it is less likely that our assembly would overestimate the amount of repetitive DNA in the genome when compared with assemblies derived from shorter reads.
Long-read sequences were assembled three times, based on three a priori assumptions of genome size (60, 65 or 71 Mbp) in order to assess the influence of these assumptions on assembly. Assuming a 60 Mbp genome resulted in an assembly of just under 60 Mbp, but when postulating either 65 or 71 Mb genomes, assemblies of long-reads resulted in 63·2 and 62·7 Mbp total length, suggesting convergence on a stable estimate in spite of differing prior assumptions. MAUVE and MUMmer alignments indicated that the alignable fractions of the T. spiralis and T. murrelli assemblies were not especially sensitive to differences in the assumed final assembly size. The smaller assembly, derived from the 60 Mbp prior, most likely resulted from excessive condensation of repetitive DNA elements. The difference between Trichinella genome assembly sizes presented here and one estimate of genome size made by flow cytometry (71 Mbp) (Mitreva and Jasmer, Reference Mitreva and Jasmer2008) may reflect as-yet undetected repetitive DNA. For example, Phobos, which can detect degenerate repeats in the genome, estimated almost twice the number of simple repeats detected by RepeatMasker; this highlights the difficulty of correctly identifying, much less assembling, degenerate repetitive DNA. If there are similar degenerative repeats among interspersed elements, ours is probably an underestimate of the true amount of repetitive DNA. It is likely that this type of repeat remains within the assemblies presented here, despite our best efforts.
The marked improvement in the assembly of T. spiralis did not significantly alter our understanding of gene content. CEGMA and BUSCO analyses suggested that all assemblies were nearly complete. Initial predictions by the Augustus and Maker gene prediction programs indicated between 10 400 and 13 500 genes are present in the long-read T. murrelli genome assembly, as compared with ~15 000 genes predicted in other studies. The presence of transposons or alternative transcripts did not account for this difference. Our gene prediction parameters may be conservative, as we found nearly 4000 fewer ‘hypothetical’ genes than were found in the 2011 T. spiralis assembly (Mitreva et al. Reference Mitreva, Jasmer, Zarlenga, Wang, Abubucker, Martin, Taylor, Yin, Fulton, Minx, Yang, Warren, Fulton, Bhonagiri, Zhang, Hallsworth-Pepin, Clifton, McCarter, Appleton, Mardis and Wilson2011) and the 2016 T. murrelli short-read assembly (Korhonen et al. Reference Korhonen, Pozio, La Rosa, Chang, Koehler, Hoberg, Boag, Tan, Jex, Hofmann, Sternberg, Young and Gasser2016). These comparisons require further analysis accompanied by RNA sequencing.
Improved organization of contigs is important for understanding genome evolution. The 2011 T. spiralis genome assembly implies that 17 genome rearrangements occurred for every Mb of sequence since its divergence from T. murrelli. Assuming an estimated 7–10 MY of divergence (Zarlenga et al. Reference Zarlenga, Rosenthal, Rosa, Pozio and Hoberg2006; Korhonen et al. Reference Korhonen, Pozio, La Rosa, Chang, Koehler, Hoberg, Boag, Tan, Jex, Hofmann, Sternberg, Young and Gasser2016), two chromosome breakages per inversion, and three breakages per translocation, 1·7–2·5 chromosomal breakages/Mbp/MY would have occurred. However, our revised assembly of T. spiralis reduces the estimate of genome breakages 10-fold, to 0·15–0·21 breakages/Mbp/MY; a rate more in line with previous estimates for the free-living nematodes Caenorhabditis elegans and Caenorhabditis briggsae (0·4–1·0/Mbp/MY) (Coghlan and Wolfe, Reference Coghlan and Wolfe2002). Furthermore, the improved organization of LCBs within the genome enables better estimates of synteny between these two species, because the syntenic correlation between single-copy orthologues (0·118 as reported by Korhonen et al. Reference Korhonen, Pozio, La Rosa, Chang, Koehler, Hoberg, Boag, Tan, Jex, Hofmann, Sternberg, Young and Gasser2016) was dependent on the 2011 arrangement of scaffolds. However, a revised estimate cannot be made until full annotation of this assembly is complete. Ongoing RNA sequencing will illuminate this issue going forward.
Concluding remarks
Trichinella murrelli and T. spiralis maintain remarkable synteny across all three chromosomes. This has enabled us to improve the assembly of each genome. Earlier hierarchical scaffolding of short reads, combined with bacterial artificial chromosomes, provided useful information for linking large numbers of contigs in the 2011 T. spiralis assembly. Here, long-read sequencing using PacBio technology substantially improved that scaffolding by spanning repeat regions and allowing more precise ordering of contigs. Together, these refined assemblies of T. murrelli and T. spiralis provide a resource for future investigations of genome conservation within the genus Trichinella, and among clade I nematodes, more generally. RNAseq studies of the gene content of these two genomes will improve comparisons between the two species and provide insight into the gene families that define the genus Trichinella and those genes that are dispensable. Furthermore, defined sets of genes may help to indicate why T. murrelli infects swine poorly and causes minimal disease whereas T. spiralis as adapted well to swine hosts and therefore provides significant danger of zoonotic disease.
GENE SEQUENCES
All sequences associated with this publication have been submitted to GenBank and can be found under:
BioProject PRJNA296806
WGS Accession LKDY00000000
Raw data can be accessed in the SRA archive: Accession numbers SRR5264263 and SRR5264264
ACKNOWLEDGEMENTS
The authors would like to thank Jonathan Shao and Brady Gaynor at USDA-ARS for bioinformatic guidance throughout the investigation.
FINANCIAL SUPPORT
Parts of this research were funded by the following grants: USDA-ARS Project 8042-32420-007-00D ‘Detection and Control of Foodborne Parasites for Food Safety’; NIFA Project 2010-004461, ‘Managing the risk of trichinellosis in organic and free range pork’; National Natural Science Foundation of China (NSFC 31520103916); Guangdong Innovative and Entrepreneurial Research Team Program (No.2014ZT05S123). P.C.T. was supported by an Oak Ridge Institute for Science and Education (ORISE) Fellowship through an interagency agreement between the U.S. Department of Energy and the USDA Agricultural Research Service.








