


INTRODUCTION
Leishmaniasis is caused by protozoan parasites of the Leishmania species that are transmitted by the bite of phlebotomine sandflies. In vertebrate hosts, Leishmania survive and multiply as non-motile amastigotes, primarily in macrophages. Amastigotes are ingested when a female sandfly takes a blood meal from an infected host. In the sandfly, amastigotes undergo a developmental programme culminating in the generation of infective metacyclic, flagellated promastigotes, which are introduced into the skin with the fly saliva at the next blood meal (Fig. 1).
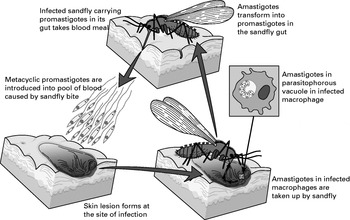
Fig 1. Schematic diagram of Leishmania life cycle.
Leishmaniasis currently threatens 350 million men, women and children in 88 countries around the world. It is estimated that 2 million new cases occur each year, with at least 12 million people presently infected worldwide (World Health Organization, Leishmaniasis Control home page: http://www.who.int/ctd/html/leish.html), and the burden of disease expressed in disability-adjusted life years (DALYs) is estimated to be almost 2 million (Table 1). The disease is endemic in Africa, Southwest Asia, the Middle East, Southern Europe, and Central and South America. Recent evidence has also shown that Leishmania-HIV co-infections are a major health problem in affected areas (Sinha, Pandey and Bhattacharya, 2005).

The genus Leishmania comprises 30 species of which about 20 are pathogenic for humans (Cupolillo et al. 2000). For most species humans are accidental hosts since leishmaniasis is primarily a zoonotic disease or has recent zoonotic origins (Ashford, 2000). The expansion of zoonotic forms of disease into areas not considered previously to be endemic, such as certain regions of the Mediterranean basin, where dogs constitute the major parasite reservoir is becoming a problem (Robertson et al. 2000).
THE DISEASE
Leishmaniasis has been traditionally classified into three major clinical entities: cutaneous, mucocutaneous and visceral (Evans, 1993).
Cutaneous leishmaniasis (CL)
The cutaneous form of the disease accounts for more than 50% of new cases of leishmaniasis. It results in formation of skin ulcers at the site of the sandfly bite, usually on exposed parts of the body, the face, neck, arms and legs. CL is caused by several species of Leishmania: L. major, L. tropica, L. aethiopica, and L. mexicana, but also by L. braziliensis, L. panamensis, L. peruviana and L. amazonensis. The disease is usually self-limiting, but the time to lesion resolution varies between species and between individuals. Some species are also noted for causing non-healing cutaneous disease. Diffuse cutaneous leishmaniasis caused by L. aethiopica, L. amazonensis and L. mexicana occurs in anergic hosts with poor immune responses. This form of disease is restricted to a few foci in Ethiopia, Kenya, Venezuela and the Dominican Republic, suggesting an important role for the genetics of the parasite as well as the genetics of the host in determining the disease phenotype. Infection is characterized by a primary lesion, which spreads to involve multiple areas of the skin with large numbers of parasites present in lesions.
Mucocutaneous leishmaniasis
In mucocutaneous leishmaniasis, usually caused by L. braziliensis, the initial skin lesions cure, but the late development of metastatic lesions can lead to partial or total destruction of the mucous membranes. Mucocutaneous leishmaniasis may also arise after inadequate treatment of some Leishmania species, and if untreated can lead to severe deformities or even death.
Visceral leishmaniasis (VL)
Visceral leishmaniasis, also known as kala-azar, is the most severe and often fatal syndrome. L. donovani, L. infantum and L. chagasi are the major species responsible for visceral leishmaniasis. They home to visceral organs and result in the pentad of syndromes comprised of fever, weight loss, splenomegaly, hepatomegaly and anaemia. If left untreated, the disease has a high mortality rate mainly due to immunosuppression and secondary infections. Some individuals develop the unusual syndrome known as post kala-azar dermal leishmaniasis (PKDL), which appears within a few years of the complete cure of VL. PKDL patients are considered a major source of parasites for new infections because of the large number of organisms in the skin accessible to sandfly bites. Visceral leishmaniasis has become a frequent infection in HIV positive individuals in endemic areas.
IMMUNOLOGY OF LEISHMANIASIS
The pathology of Leishmania infection is determined not only by the parasite species, but also by host genetics and immune factors. Most of the experimental immunological data come from mouse models and less is known about the immunology of human leishmaniasis. Although mouse models have been used for the study of both cutaneous and visceral leishmaniasis, they more closely reflect the situation in human cutaneous leishmaniasis than visceral disease.
In the case of cutaneous leishmaniasis, effective protection against infection has been largely attributed to the development of a potent CD4+ Th1− type immune response, characterized by the production of IL-12 and IFN-γ, which subsequently mediates macrophage activation, nitric oxide production and parasite killing (Rogers et al. 2002; Alexander and Bryson, 2005; Coler and Reed, 2005). A clear-cut polarisation of T helper cell responses is not evident in human leishmaniasis which shows a mixed Th1 and Th2 immune response (von Stebut and Udey, 2004). The ability of the infected individual to mount a Th1 response is considered to be partially responsible for the observed differences in the clinical picture of leishmaniasis. However, Th2 cell mediated responses have not been unequivocally associated with a failure to mount a protective response, and therefore causing long-lasting cutaneous or systemic infection. The disease phenotype may be attributed to the Leishmania species causing the disease (McMahon-Pratt and Alexander, 2004).
In recent years, the involvement of CD8+ T cells has also been shown to play an important role in immunity against cutaneous leishmaniasis (Belkaid et al. 2002; Rodrigues et al. 2003). Similarly, innate immunity, including natural killer cells, IL-1α and myeloid differentiation factor 88 (MyD88) act as immunomodulators determining early resistance to infection (Handman, 2001). Surprisingly, wound healing and tissue repair mechanisms have recently been implicated in resistance to cutaneous leishmaniasis (Sakthianandeswaren et al. 2005).
Recent studies on the generation and maintenance of central memory (CM) and effector memory (EM) CD4+ T cells during cutaneous Leishmania infection shed new light on the design of effective vaccination strategies against Leishmania (Gollob, Antonelli and Dutra, 2005; Scott, 2005). In the murine model of disease, it has been suggested that the constant presence of live parasites is required for maintaining EM CD4+ T cells, but might not be essential for the maintenance of CM CD4+ T cells (Seder and Sacks, 2004; Zaph et al. 2004). Thus, the efficacy of killed or subunit vaccines might be greatly enhanced by using adjuvants that favour the generation of CM CD4+ T cells. However, the importance of persistent infection for maintaining an effective long-lasting protective response is controversial (Belkaid et al. 2002; Uzonna et al. 2001). Since vaccines need to generate immunological memory, a better understanding of the formation and maintenance of CM and EM CD4+ T cells in both animal models and human disease will be critical for their development.
TOWARDS A VACCINE
Current treatment of leishmaniasis primarily relies on chemotherapy, with some attempts at using immunotherapy (Ghosh et al. 2003; Santos et al. 2003; Borja-Cabrera et al. 2004). The first line of treatment is predominantly based on pentavalent antimonials, a classic treatment in most of the endemic areas. However, its usefulness has been compromised by the emergence of resistance. The second line treatment includes drugs such as amphotericin B and pentamidine, which are characterized by high efficacy, but are relatively expensive and have severe side effects (Berman, 2003; Davis and Kedzierski, 2005). Newer drugs, such as the lipid formulations of amphotericin B have been effective in the treatment of visceral leishmaniasis, but the prohibitive cost of this drug means that it is unavailable to the majority of patients (Berman et al. 1998; Murray, 2004). Recently, miltefosine was shown to be an effective oral treatment for visceral leishmaniasis in India (Sundar et al. 2002), and for cutaneous leishmaniasis in South America (Soto et al. 2001).
Leishmaniasis in general, but particularly cutaneous leishmaniasis, is probably one of a few parasitic diseases that is most likely to be controlled by vaccines. The relatively uncomplicated leishmanial life cycle and the fact that recovery from a primary infection renders the host resistant to subsequent infections indicate that a successful vaccine is feasible. Extensive evidence from studies in animal models, mainly mice, indicates that solid protection can be achieved upon immunisation with defined protein or DNA vaccines.
Attempts of vaccination, more appropriately defined as controlled infection, against cutaneous leishmaniasis can be traced back hundreds of years. Based on the observation that cutaneous leishmaniasis usually involves a benign ulcer which heals spontaneously and is accompanied by protection from reinfection, the ancient Middle Easterners started the practice of deliberately exposing uninfected individuals to sandfly bites or to infectious material from lesions (Handman, 2001). This strategy of controlled infection is still being tested in certain areas today, though in a more standardised manner (Khamesipour et al. 2005). During the past several decades, extensive efforts have been made to search for an effective Leishmania vaccine. Vaccine formulations including killed, live attenuated parasites, recombinant Leishmania proteins or DNA encoding leishmanial proteins, as well as immunomodulators from sandfly saliva have been examined (Table 2). Although to date, there is no vaccine against Leishmania, several of the vaccine preparations are at advanced stages of clinical testing.
An ideal anti-leishmanial vaccine would need to possess several attributes, but not all of them may be easily achievable. These include; (1) Safety; (2) Affordability to the populations in need; (3) Induction of CD4+ and CD8+ T cell responses and long-term immunological memory that can be boosted by natural infections, thus minimising the number of immunisations; (4) Effectiveness against species causing CL and VL; (5) Stability at room temperature eliminating the need for a cold chain to preserve potency and; (6) Effectiveness as a prophylactic as well as a therapeutic vaccine. While the cost-effectiveness and safety issues can be relatively straightforward to resolve, the induction and maintenance of the required immune responses are much more difficult to solve and cross-species protection may not be achieved by the same vaccine.
Leishmanisation
Leishmanisation, the inoculation of live virulent Leishmania, has been practised for over a century (Wenyon, 1911; Greenblatt, 1988). The injection of viable parasites produces presumably controlled lesion and induces T cell mediated immunity (Nadim et al. 1983). Leishmanisation was used successfully for a long time in the republics of the former Soviet Union, Israel and Iran. However, this practice has been abandoned in most countries mainly due to safety issues. Some individuals vaccinated with the virulent parasites developed long-term large lesions that did not heal and required medical treatment. In rare cases, exacerbated chronic skin disease or even immunosuppression have been reported. A different type of problem has been the loss of virulence after repeated in vitro passage of the parasites, which made the standardisation of the vaccine difficult.
The traditional practice of leishmanisation has recently made a comeback in certain endemic regions, mainly because to date, it is the only vaccine against Leishmania with proven efficacy in humans (Tabbara et al. 2005). Efforts are being made to improve safety of leishmanisation by the inclusion of drug-sensitive Leishmania mutants with suicide genes for controlled infection (Muyombwe et al. 1997; Davoudi et al. 2005), inclusion of killed parasites to reduce the size and duration of lesions, or by using adjuvants that promote more rapid onset of anti-leishmanial immunity and swift healing of lesions (Khamesipour et al. 2005; Tabbara et al. 2005).
Killed vaccines
Killed parasite vaccines have been proposed as both prophylactic and therapeutic vaccines. The therapeutic application may be particularly important in cases of drug resistant refractory disease. However, the whole-cell, killed vaccines have been rather poorly defined and variable in potency, hence they have rendered inconclusive results. Vaccination with killed parasites dates back to the late 1930s and was pioneered by Brazilian scientists. The vaccine was based on cultured, killed promastigotes and resulted in a reduction of the number of leishmaniasis cases in the populations under study. However, it was abandoned probably due to the low incidence of leishmaniasis in endemic areas where it was undergoing testing (Marzochi et al. 1998). Later vaccination studies by Mayrink and colleagues in new areas confirmed previous reports, but could not be properly evaluated due to the disappearance of leishmaniasis from one area under study and population movement (Mayrink et al. 1979, 1985). In these studies, a vaccine containing promastigotes of five killed Leishmania strains was shown to be safe and immunogenic as measured by the leishmanin skin test (LST) conversion, but conferred only a small degree of protection (50%). The production of a vaccine known as Leishvacin containing only one of the initial strains (L. amazonensis, IFLA/BR/67/PH8) was subsequently initiated (Botelho et al. 1998; Marzochi et al. 1998). Phase III clinical trials in Ecuador and Colombia showed that the L. amazonensis vaccine was safe, but not efficacious (Armijos et al. 2004; Velez et al. 2005). Most cases of leishmaniasis in Colombia and Ecuador are caused by species of the subgenus Viannia. It has been argued that vaccination using these killed parasites might only protect against homologous infection. The observation that vaccinated individuals developed a Th1 immune response without being protected against infection is in agreement with several similar observations in mice and vervet monkeys (Sjolander et al. 1998a; Gicheru et al. 2001), and suggests that the induction of a Th1 immune response may be necessary, but not sufficient for protection against cutaneous leishmaniasis.
Convit and colleagues (Sharples et al. 1994) pioneered the use of immunotherapy with a combination of killed L. amazonensis promastigotes and BCG for the treatment of localised cutaneous leishmaniasis. In Venezuela, a clinical healing rate of more than 95% was achieved and cure was associated with a Th1-like immune response in the patients (Cabrera et al. 2000; Convit et al. 2003).
A modified form of the vaccine using pasteurised L. braziliensis promastigotes and live BCG was effective in the treatment of refractory mucocutaneous leishmaniasis and early cases of diffuse cutaneous leishmaniasis (Convit et al. 2004). In Brazil, the combination of killed L. amazonensis promastigotes with a half-dose regimen of meglumine antimoniate was also shown to be highly effective for the treatment of cutaneous leishmaniasis (Machado-Pinto et al. 2002).
A longitudinal epidemiological study of L. donovani infection in eastern Sudan suggested that previous exposure or infection with L. major protected against visceral leishmaniasis caused by L. donovani (Zijlstra et al. 1994). With this in mind, a vaccine consisting of autoclaved L. major and BCG was used and shown to be safe, and significantly, it induced IFN-γ production in healthy volunteers (Satti et al. 2001). Alum precipitation of L. major improved the immunogenicity of the vaccine and induced a strong delayed type hypersensitivity (DTH) reaction in volunteers (Kamil et al. 2003). Successful protection against L. donovani infection has been achieved with killed L. major in simian, canine and hamster models (Misra et al. 2001; Srivastava et al. 2003; Mohebali et al. 2004) and provides the basis for further human trials.
Live-attenuated vaccines
Vaccination with attenuated parasites, which are infectious but not pathogenic, has major advantages compared to leishmanisation or vaccination with killed promastigotes. Attenuated parasites are taken up by the natural host cell into the same compartment as the virulent organisms and persist long enough for the induction of the appropriate immune response without causing disease.
Attenuated Leishmania vaccines have been produced by long-term culture, by culturing under gentamicin pressure, irradiation, chemical mutagenesis and, more recently, by deleting genes from the Leishmania genome. Parasites lacking genes essential for long-term survival in the mammalian host, such as the gene encoding dihydrofolate reductase-thymidylate synthetase (DHFR-TS) were tested as potential vaccines (Titus et al. 1995; Veras et al. 1999). DHFR-TS-deficient L. major parasites induced limited protection against infection with L. major and L. amazonensis in a murine model. However, vaccination with L. major lacking DHFR-TS failed to show protective immunity in a primate model (Amaral et al. 2002). The limited protection conferred by these attenuated parasites might be due to their rapid elimination by the host. Targeted deletion of other virulence genes, such as the cysteine proteinase genes or the lpg1 gene, encoding a putative galactofuranosyl transferase involved in the biosynthesis of the virulence factor, lipophosphoglycan, resulted in parasites which, although attenuated, could still cause disease, making such parasite lines unacceptable as vaccines (Alexander, Coombs and Mottram, 1998; Huang and Turco, 1993; Ryan et al. 1993). Recently, L. major parasites lacking the lpg2 gene encoding an enzyme involved in the transport of GDP-mannose to the Golgi apparatus, were shown to persist in BALB/c mice without causing lesions, and to protect against homologous infection (Uzonna et al. 2001). However, in some experiments these mutants could regain their ability to cause infection through a compensatory mechanisms (Spath et al. 2004). Moreover, persistence was not sufficient to confer immunity in C57BL/6 mice. Co-administration of CpG oligonucleotides was necessary for protection against L. major infection (Kebaier et al. 2006). Surprisingly, the protection induced by those attenuated organisms was not associated with a significant Th1 response as measured by IFN-γ production and delayed type hypersensitivity (DTH), suggesting that the Th1-like responses might not always be essential or correlate with protective immunity.
It is of interest that L. tarentolae, which is not pathogenic in humans, was shown not only to activate dendritic cell maturation and induce Th1 type immune response in mice, but to protect BALB/c mice against an infectious challenge with L. donovani (Breton et al. 2005).
Molecularly defined vaccines against cutaneous leishmaniasis
The majority of the vaccine development effort has been directed towards the cutaneous form of the disease caused by L. major. The murine model of cutaneous leishmaniasis, which mimics many aspects of the human disease and also allows the dissection of the role of cytokines and T helper responses, has been used as a tool for assessing vaccine candidates. One caveat is the fact that the precise immune mechanisms underlying human cutaneous leishmaniasis are still not fully understood, and the responses necessary for protection by vaccination are also not as clear as in the mouse model (Louis et al. 2002; Gumy, Louis and Launois, 2004).

Early vaccination studies indicated that immunisation with the complex surface glycolipid ligand for the host cell receptor, lipophosphoglycan (LPG), provided protection against cutaneous leishmaniasis caused by L. major (Handman and Mitchell, 1985; McConville et al. 1987). Protection with LPG was heavily dependent on the integrity of the molecule as the water-soluble PG lacking the GPI anchor had exacerbatory rather than protective properties (Mitchell and Handman, 1986). More recently, studies with L. donovani-derived LPG suggested that it might be a promising candidate for development of a transmission blocking vaccine (Tonui et al. 2001). However, immunisation with L. donovani LPG showed that experimental animals were not protected against homologous (Tonui et al. 2003) or heterologous challenge (Tonui, 2003), despite Th1 immune responses being observed. These results may be explained by the fact that a water-soluble form of LPG was used in these studies, which had been previously shown to lack protective efficacy against the cutaneous form of disease.
VACCINATION WITH RECOMBINANT DNA-DERIVED PROTEINS
gp63 – leishmanolysin
The most comprehensively studied anti-leishmanial vaccine candidate is the surface-expressed glycoprotein gp63, or leishmanolysin. Gp63 is a zinc metalloprotease, abundantly expressed on the promastigote surface. It binds to the macrophage complement receptor 3 (CR3) mediating internalisation of promastigotes (Russell and Wright, 1988; Handman, 1999). The polypeptide has been produced in a variety of expression systems (bacterial, viral or mammalian) (Handman, 2001) and used extensively in vaccination and immunological studies. When used as a recombinant protein expressed in E. coli, gp63 either failed to protect mice against L. major infection (Handman, Button and McMaster, 1990) or offered only partial protection in monkeys (Olobo et al. 1995). In contrast, immunisation with the native protein purified form L. major led to protection of mice against challenge with either L. mexicana or L. major (Russell and Alexander, 1988; Rivier et al. 1999). Successful vaccination was also achieved using gp63 expressed in BCG (Connell et al. 1993; Abdelhak et al. 1995) and attenuated Salmonella (Yang et al. 1990; Xu et al. 1995; McSorley, Xu and Liew, 1997; Gonzalez et al. 1998). These promising findings were overshadowed by variable (mostly negative) T cell responses in humans (Jaffe et al. 1990; Kemp et al. 1991; Mendonca, Russell and Coutinho, 1991; Russo et al. 1991) and the inability of T cells from mice immunised with gp63 to elicit protective responses in macrophages (Rivier et al. 1999). Nonetheless, researchers focused their efforts on gp63 DNA-based vaccines (described below) and gp63-derived synthetic peptides. The latter were successfully tested in an animal model of cutaneous leishmaniasis and triggered long-lasting T cells responses (Jardim et al. 1990; Spitzer et al. 1999).
With the increased understanding of the importance of dendritic cells in the induction of T cell responses, synthetic peptides as well as gp63 itself were used to pulse dendritic cells (DC) in order to investigate the potential of a DC-based subunit vaccine (Berberich et al. 2003; Tsagozis, Karagouni and Dotsika, 2004). Antigen-pulsed DCs were able to protect mice against infection, and protection was associated with a shift towards a Th1 response (Flohe et al. 1998). More detailed studies showed that mice vaccinated with DCs loaded with leishmanolysin were capable of controlling infection with L. major and the degree of protection correlated with the level of IL-12 expression and Th1 response (Berberich et al. 2003). However, the selection of antigen used for pulsing the DCs is critical, since some gp63-derived synthetic peptides led to protection while others led to exacerbation of disease (Tsagozis et al. 2004). In another study, gp63 was co-expressed with human CD40L, a potent inducer of IL-12 production (Chen, Darrah and Mosser, 2001). Vaccination with cells expressing both molecules partially protected C57BL/6 mice against L. amazonensis infection. In contrast, earlier reports indicated that E. coli-derived gp63 co-administered with IL-12 failed to protect BALB/c mice against L. major infection suggesting an important role for host genetics for the ability to turn on protective immune responses to the vaccine.
Gp63 has been reported to be a major component of the Leishvacin vaccine tested in healthy volunteers in Brazil (Nascimento et al. 1990). The Leishvacin components have been identified (Cardoso et al. 2003) and their protective efficacy was tested in mice using different combinations with gp63 (Mora et al. 1999). Gp63 was also the immunodominant component of the L. donovani mixture of promastigote proteins encapsulated in liposomes and tested for protection against experimental visceral leishmaniasis (Afrin et al. 2002).
gp46/M2/Parasite Surface Antigen 2
Another vaccine candidate has been a GPI-anchored membrane protein gp46/M-2 or Parasite Surface Antigen 2 (PSA-2), that belongs to a gene family present in all Leishmania species bar L. braziliensis (Jimenez-Ruiz et al. 1998; Murray, Spithill and Handman, 1989; Lohman, Langer and McMahon-Pratt, 1990; McMahon-Pratt et al. 1992). PSA-2 consists of several leucine-rich repeat (LRRs) motifs that are shared with an unrelated surface proteophosphoglycan (PPG) (Montgomery et al. 2000). PSA-2 is involved in macrophage invasion through the interaction of its LRRs with CR3 (Kedzierski et al. 2004). There are three distinct L. major PSA-2 polypeptides expressed on the promastigote surface, but only one on amastigotes (Symons et al. 1994). There is a great deal of similarity between PSA-2 proteins from different species, but there is a high degree of heterogeneity between different members of the family (Murray and Spithill, 1991; Symons et al. 1994). Immunisation with the three polypeptides of native promastigote PSA-2 protected mice against L. major infection (Handman et al. 1995), but vaccination with a recombinant E. coli-derived promastigote or amastigote protein showed lack of protective efficacy despite the ability to induce Th1 polarised responses (Handman et al. 1995; Sjolander et al. 1998a). Immunisation with L. major PSA-2 expressed episomally in L. mexicana promastigotes conferred protection against cutaneous leishmaniasis in mice (Handman et al. 1995). Protective vaccination was also achieved against L. amazonensis (Champsi and McMahon-Pratt, 1988; McMahon-Pratt et al. 1993). In a recent study, protection of susceptible BALB/c mice by vaccination with secreted/excreted L. major antigens was attributed to a combination of PSA-2 and LPG contained in the preparation (Tonui et al. 2004). However, the synergistic or additive effect of other molecules in the mix could have enhanced the protective efficacy of PSA-2.
LACK/p36, Leishmania homologue for the receptors of activated C kinase
The Leishmania homologue for receptors of activated C kinase (LACK) is a conserved antigen expressed in both leishmanial life cycle stages (Mougneau et al. 1995). The protein belongs to the family of WD 40 repeat proteins restricted to eukaryotes and is involved in several regulatory functions. LACK is localised in the cytoplasm, bound to multiprotein complexes involved in DNA replication and RNA synthesis (Gonzalez-Aseguinolaza et al. 1999). Immunisation with LACK appears to promote the expansion of IL-4 secreting T cells and thus skewing the response towards deleterious Th2 responses (Launois et al. 1997; Julia and Glaichenhaus, 1999). Susceptible BALB/c mice that were made tolerant to LACK had diminished early Th2 responses and were able to develop protective Th1 responses leading to the control of L. major infection (Julia, Rassoulzadegan and Glaichenhaus, 1996).
Despite its propensity to induce Th2 type immune responses, immunisation with recombinant, truncated LACK co-administered with IL-12 conferred protection against cutaneous infection in mice (Mougneau et al. 1995), and recombinant LACK and IL-12 triggered short term protective responses, but failed to elicit long-term immunity (Gurunathan et al. 1998). The prime/boost immunisation with vaccinia virus expressing LACK led to protective immune responses and partial protection in homologous (Gonzalo et al. 2001) and heterologous (Gonzalo et al. 2002) challenge systems. Since co-administration of IL-12 appeared to be an essential component of LACK vaccination, a vaccine trial was conducted using LACK expressing Listeria monocytogenes as a delivery system. L. monocytogenes is a known inducer of IL-12 production and immunisation with recombinant bacteria induced Th1 responses and partial protection against L. major challenge (Soussi et al. 2000). So far, the protective efficacy of LACK has been mainly demonstrated in the L. major model. Surprisingly, despite its conservation amongst the Leishmania species, LACK failed to protect against visceral leishmaniasis, although immunisation induced strong Th1 responses (Melby et al. 2001). This observation highlights the differences between various types of leishmaniasis and points to the distinct requirements for protection that may be expected from a vaccine against CL and VL.
dp72 and P0
In contrast to the strictly L. major species-specific protection with LACK, cross-species protective efficacy has been demonstrated for some antigens such as dp72. This protein, purified from L. donovani promastigotes was able to protect mice against L. major challenge (Rachamim and Jaffe, 1993). Cross-species protection is also a feature of the acidic ribosomal P0 protein from L. infantum that was able to protect C57BL/6 from L. major infection. In this case, however, protection could not be induced in BALB/c mice, reinforcing the importance of host genetics not only in determining susceptibility to infection, but also in the ability to induce protection by vaccination (Iborra et al. 2005).
Amastigote cysteine proteases
Antigens expressed in the amastigote may be the most important vaccine candidates since the mammalian form of the parasite is both the main inducer and the target of the immune response. The cathepsin L-like cysteine proteinases (CPs) belonging to the papain superfamily are thought to be good vaccine candidates due to their high immunogenicity and important role in host-parasite interaction (Wolfram et al. 1995; Rafati, Fasel and Masina, 2003). Three classes of CPs have been identified; Type I (CPB), Type II (CPA) and Type III (CPC) (Robertson et al. 1996). The CPA and CPB are targeted by the immune system as demonstrated by reactivity of human sera from individuals who either recovered or showed clinical symptoms of cutaneous leishmaniasis (Rafati et al. 2001). Immunisation of mice with recombinant cysteine proteinase induced high production of IFN-γ and offered partial protection against L. major challenge (Rafati et al. 2000). More detailed vaccination studies with CPB and CPA demonstrated that only recombinant CPB, but not CPA was able to trigger immune responses that partially protected experimental animals against challenge, and protection depended on IFN-γ producing CD8+ T cells (Rafati et al. 2002). Recently, a hybrid fusion protein composed of CPA and CPB was used to immunise mice and partial protection against L. major infection was obtained (Zadeh-Vakili et al. 2004). Protection was similar to that obtained by immunisation with CPB alone (Rafati et al. 2002), but much higher levels of IFN-γ were detected upon immunisation with the hybrid protein, pointing towards a dominant Th1 response.
In other leishmanial species, a homologue of cysteine proteinase (A2) from L. pifanoi partially protected mice against homologous challenge (Soong et al. 1995), and inclusion of recombinant L. mexicana cysteine proteinase 5 in a cocktail with gp63 and acid phosphatase triggered protective responses in C57BL/6 mice, but not BALB/c or CBA/J (Aebischer et al. 2000). The protective effect of CPs has also been assessed in visceral leishmaniasis (VL) in the canine model (Rafati et al. 2003; Nakhaee et al. 2004). Immunisation with CPB and CPA from L. infantum delivered as a combination of DNA and protein showed good efficacy (Rafati et al. 2005).
Amastigote P4 and P8
Other amastigote-specific vaccine candidates include membrane proteins P4 and P8, which conferred protection against L. pifanoi challenge (Soong et al. 1995). P4 is a membrane-associated, single strand-specific nuclease present in all Leishmania species (Kar et al. 2000). In T cell proliferation assays, P4 selectively elicited Th1-like responses in peripheral blood mononuclear cells from patients infected with L. braziliensis (Haberer et al. 1998). CD4+ T cells producing IFN-γ, lymphotoxin and macrophage migration inhibitory factor played a major role in protection in animals vaccinated with P4 (Kar, Metz and McMahon-Pratt, 2005). The function of P8 is not known, but the L. pifanoi polypeptide conferred protection in a heterologous challenge system against L. amazonensis (Soong et al. 1995), and triggered T cell recall responses in patients with L. braziliensis and L. amazonensis infections similar to P4 (Silveira et al. 1998).
VACCINATION AGAINST VISCERAL LEISHMANIASIS
Although the demands from a VL vaccine are more complex than for a CL vaccine, it is believed that human VL trials will follow any successful CL immunisation programme. Whether the same vaccine will work against both forms of the disease remains to be seen. Similar to the situation in cutaneous leishmaniasis, protection against VL correlates with production of IFN-γ by Th1-type cells. However, co-existence of Th1 and Th2 responses has been reported in VL patients as well as experimental animals. In contrast to CL, Th2 responses do not hinder Th1 responses and early Th2 cytokines may in fact enhance IL-12 and IFN-γ production later on. Moreover, humoral immune responses seem to play a role in anti-VL immunity. It appears that a vaccine against visceral leishmaniasis may need to generate both cellular and humoral immune responses (Ravindran and Ali, 2004).
VL vaccination studies have been hampered by the lack of a suitable animal model of disease. The best animal models are the natural combination of dogs and L. infantum or L. chagasi (Hommel et al. 1995) and L. donovani in golden hamsters (Requena et al. 2000). Several clinical symptoms and pathogenic features of infection in both models are similar to the human disease. The canine model is particularly useful in evaluating vaccine candidates since successful vaccination of dogs is thought, at least to some extent, to control the spread of disease to humans in endemic areas where the dog is the reservoir of infection (Tesh, 1995). However, both models which use outbred animals also suffer from lack of immunological reagents and assays needed for the dissection of immune responses.
The mouse model of VL has been the most widely used system. It has the advantage that there are many different knockout mice with specific lesions in the immune system and there are good immunological reagents. However, it does not fully reproduce the disease observed in humans. The Th1 and Th2 polarisation has not been observed for L. donovani and often the mice have to be injected intravenously with large numbers of amastigotes in order to achieve visceral disease (Ravindran and Ali, 2004).
Single antigen vaccines
The protective efficacy of several antigens delivered either as DNA vaccines or subunit vaccines has been tested in the canine model of visceral leishmaniasis. Early studies showed that dp72 protected mice against L. donovani infection (Jaffe, Rachamim and Sarfstein, 1990; Rachamim and Jaffe, 1993). Despite these early successes, there has been no progress on the use of this antigen for the development of vaccines. A handful of other recombinant proteins have been tested against visceral leishmaniasis in murine models. The LACK DNA vaccine was tested in dogs and mice with variable outcomes (Melby et al. 2001; Ramiro et al. 2003; Dondji et al. 2005; Marques-da-Silva et al. 2005).
The L. donovani amastigote LCR1 protein containing 67-amino acid repeats homologous to repeats in a Trypanosoma cruzi flagellar polypeptide, was administered as recombinant protein or expressed in BCG and tested for protection in mice. The recombinant protein induced partial protection against L. chagasi challenge (Wilson et al. 1995). Immunisation with BCG-LCR1 elicited better protection than the protein alone, but protection depended on the site of immunisation, subcutaneous delivery being better than intra-peritoneal (Streit et al. 2000).
Immunisation with the A2 cysteine proteinase delivered as recombinant protein or as DNA offered protection against invasion of macrophages and disease progression (Ghosh, Labrecque and Matlashewski, 2001; Ghosh, Zhang and Matlashewski, 2001).
Recombinant hydrophilic acylated surface protein B1 (HASPB1), a member of a family of proteins expressed only in metacyclic and amastigote stages of development of several Leishmania species, was protective in the mouse model of VL and interestingly, protection did not require any adjuvants and seemed to be generated via mechanisms reminiscent of DNA vaccination (Stager, Smith and Kaye, 2000).
The PapLe22 antigen, a protein of unknown function, which localises to the promastigote nucleus is recognised by T cells from visceral leishmaniasis patients (Suffia et al. 2000). Although PapLe22 DNA vaccination led to a marked decrease in parasite burden in immunised hamsters (Fragaki et al. 2001), it induced IL-10 production in peripheral blood mononuclear cells from visceral leishmaniasis patients indicating that in humans it might actually contribute to pathogenesis (Suffia et al. 2000). Therefore, its use as a vaccine would need to consider the possibility that it may exacerbate disease. PapLe22 vaccine may be able to protect if the vaccine formulation would redirect T cell responses towards Th1 type responses.
The leishmanial antigen ORFF, also a protein of unknown function (Ghosh et al. 1999), was able to induce protective immunity against L. donovani challenge when administered with CpGs oligonucleotides (Tewary et al. 2004).
Poly-protein vaccines
Apart from defined single molecules, multicomponent vaccines have been demonstrated to afford protection against VL in experimental animals. Recombinant Q protein formed by fusion of antigenic determinants from four cytoplasmic proteins from L. infantum (Lip2a, Lip2b, P0 and histone H2A) co-administered with live BCG protected 90% of immunised dogs by enhancing parasite clearance (Molano et al. 2003).
DNA vaccines
In 1995, DNA vaccination was proposed to be the way of the future (Waine and McManus, 1995). DNA vaccines are relatively simple to produce and administer, they are often very immunogenic and offer a protein that is usually correctly folded and may be post-translationally modified in a fashion similar to the native protein. Such vaccines are able to elicit humoral, CD4+ and CD8+ T cell immune responses, which can be further modulated by the addition of cytokines and/or CpG oligonucleotides (Alarcon, Waine and McManus, 1999; Restifo et al. 2000). They can also be modulated by prime-boost strategies that involve priming with DNA and boosting with protein (McShane, 2002).
Most nucleic acid vaccination efforts have been directed against viral infections, which require induction of CTL responses, a major feature of DNA vaccines. This method of immunisation is also attractive for leishmaniasis since the induction of Th1 responses is also a general property of DNA vaccines (Gurunathan, Klinman and Seder, 2000). In addition, a growing body of evidence implicates CD8+ T cells in anti-leishmanial immunity (Rodrigues et al. 2003). Most of the antigens described in the previous sections and delivered as recombinant proteins or expressed in live, microbial delivery systems have also been tested as DNA vaccines.
The gene encoding gp63 was the first to be used as a DNA vaccine, and immunised mice developed strong Th1 responses as well as significant resistance to infection with L. major (Xu and Liew, 1994, 1995). In another study, 30% protection was reported in immunised mice, with indications of strong Th1 responses being elicited by vaccination (Walker et al. 1998). More recently, a comparative study evaluating different DNA vaccine candidates including gp63 showed that protection was transient, and eventually the immunised mice developed lesions similar to those observed in controls (Ahmed et al. 2004). The same study also included PSA-2, which did not confer protection. This is in contrast with previous studies using PSA-2 DNA immunisation as either prophylactic (Sjolander et al. 1998b) or therapeutic vaccines (Handman et al. 2000), which showed protection associated with strong Th1 responses. The difference in outcome between the two studies could be due to the use of susceptible BALB/c mice in the first, and resistant C3H/He mice in the second. Another comparative study demonstrated that gp63 DNA immunisation was able to reduce lesion size as well as parasite burden, while gp46/PSA-2 DNA vaccination led only to a reduction in lesion size without reduction of parasite burden (Dumonteil et al. 2003).
LACK is the most extensively studied DNA vaccine against both cutaneous and visceral leishmaniasis. DNA vaccination with a plasmid harbouring the LACK gene with, or without co-administration of IL-12 induced robust, long-lasting protection against L. major challenge in mice, dependent on the immunoregulatory role of CD8+ T cells (Gurunathan et al. 1997, 1998, 2000). In a heterologous challenge system, priming with L. infantum LACK followed by a booster with vaccinia virus expressing LACK afforded protection against L. major infection (Gonzalo et al. 2002). The protection was further enhanced by co-administration of plasmids expressing IL-12 and IL-18 cytokines (Tapia et al. 2003). Since previous studies showed that LACK-induced immunity was dependent on CD8+ T cells, boosting with vaccinia virus probably enhanced this immunity by expanding the CD8+ T cells population (Zavala et al. 2001). Boosting with recombinant Salmonella expressing LACK following a priming injection with DNA also conferred protection against infection and skewed responses towards Th1, thus enhancing the protection observed upon immunisation with DNA or Salmonella alone (Lange et al. 2004). The prime-boost regimen was also employed to immunise dogs against visceral leishmaniasis and elicited protective responses in 60% of vaccinated animals (Ramiro et al. 2003). Protective vaccination against L. major was also achieved following delivery of LACK in a minimalistic, immunogenically defined gene expression (MIDGE) vector (Lopez-Fuertes et al. 2002) with lower doses of plasmids required for protection. The intranasal delivery of LACK DNA also protected mice against L. amazonensis challenge (Pinto et al. 2004). These positive outcomes are overshadowed by several studies where immunisation with LACK offered no protection. These reports are mainly restricted to visceral leishmaniasis, but there are also reports in the L. major (Ahmed et al. 2004) and L. mexicana models of disease (Dumonteil et al. 2003).
Melby and colleagues (Melby et al. 2001) reported that despite triggering strong Th1 responses the LACK DNA vaccine did not induce protection in mice against L. donovani challenge. Moreover, the co-administration of IL-12 did not improve the protective outcome. A recent study in the L. chagasi model, confirmed that LACK DNA vaccination does not confer protection against VL despite the presence of Th1 responses (Marques-da-Silva et al. 2005).
Several other antigens have been successfully tested as DNA vaccines against cutaneous or visceral infection. The former group include acidic ribosomal protein P0 (Iborra et al. 2003), P4 nuclease (Campbell et al. 2003) and paraflagellar rod protein 2 (PRP-2) (Saravia et al. 2005), whereas the latter contains ORFF (Sukumaran et al. 2003; Tewary et al. 2005), kinetoplastid membrane protein-11 (KMP-11) (Basu et al. 2005), CPA and CPB (Rafati et al. 2005) and NH36, a main component of the fucose-mannose ligand (Aguilar-Be et al. 2005).
DNA vaccination against Leishmania is considered a promising technology, but no development of such a vaccine for use in humans has been reported so far. Conflicting reports as to the protective efficacy of the antigens delivered in this mode add to the confusion in the field. To complicate issues further, protective outcomes seem to be influenced by many factors including plasmid backbone, number of injections, challenge dose and virulence of the leishmanial strain, developmental stage of the parasite (promastigote vs amastigote), experimental protocol employed, immunomodulators and type of animal model. Therefore, it is not surprising that the initial enthusiasm has been tempered by the complexities and difficulties that have surfaced.
THE SECOND GENERATION LEISHMANIA VACCINES
The best protection against leishmaniasis has been obtained after recovery from natural infection or following deliberate leishmanisation. In contrast, the first generation vaccines using immunisation with crude parasite preparations or killed Leishmania stocks resulted in variable protection ranging from good to no protection at all (Antunes et al. 1986; Mayrink et al. 1986; Genaro et al. 1996; Sharifi et al. 1998; De Luca et al. 1999, 2001; Khalil et al. 2000). Similarly, outcomes of animal vaccination trials with defined molecules or DNA vaccines have been variable, as outlined above. The thinking behind the second generation vaccines is that an effective anti-leishmanial vaccine needs to mimic as many aspects as possible of the types of antigens and the microenvironment from which the antigens are delivered during natural infection. The vaccine should also consider some practical aspects. For example, the vaccine should be delivered as a single, defined molecule to facilitate compliance with regulatory and manufacturing standards and to lower the overall production costs. Ideally, the vaccine should protect against cutaneous as well as visceral leishmaniasis.
To date, a promising second-generation vaccine, Leish-111f has been engineered and assessed in Phase I clinical trials in healthy volunteers (Reed and Campos-Neto, 2003a; Coler and Reed, 2005). Leish-111f is a single polyprotein composed of three molecules fused in tandem; the L. major homologue of eukaryotic thiol-specific antioxidant (TSA) (Webb et al. 1998), the L. major stress-inducible protein-1 (LmSTI1) (Webb et al. 1997) and the L. braziliensis elongation and initiation factor (LeIF) (Skeiky et al. 1998). The selected proteins are expressed in promastigotes and amastigotes across the Leishmania genus and have been shown to afford protection in the mouse model when administered as single antigens or as a combination (Campos-Neto et al. 2001, 2002; Coler et al. 2002; Mendez et al. 2001; Rhee et al. 2002; Skeiky et al. 2002; Webb et al. 1996, 1998). Initial immunisation trials in mice demonstrated that Leish-111f was able to protect mice against L. major and L. amazonensis infection (Coler et al. 2002; Skeiky et al. 2002). Upon immunisation with Leish-111f responses to its individual components were maintained and the protection was equal or better than that obtained by the administration of a mix of individual components. A crucial component of the Leish-111f vaccination programme is the adjuvant. The vaccine is efficacious when co-administered with IL-12, as well as a detoxified derivative of the lipid A from the lipopolysaccharide of Salmonella minnesota formulated with squalene (MPL-SE) that has been approved for use in humans (Reed, Coler and Campos-Neto, 2003b). Leish-111f is to be initially tested as a therapeutic vaccine against cutaneous leishmaniasis.
There is some evidence that the Leish-111f vaccine can also induce partial protection against visceral leishmaniasis in animal models (Reed et al. 2003b). Although recent reports have suggested that the mixture of the recombinant components of Leish-111f was highly immunogenic in dogs (Fujiwara et al. 2005), Leish-111f failed to protect dogs against infection and did not prevent disease development in a recent Phase III vaccine trial in dogs (Gradoni et al. 2005). A high percentage (95%) of the vaccinated dogs showed evidence of L. infantum infection.
USE OF DENDRITIC CELLS FOR VACCINE DELIVERY
Over the last few years, insights into the role of the innate immune system, in particular dendritic cells (DC), in the initiation and development of anti-leishmanial immunity, have provided the impetus for the use of dendritic cells to deliver vaccine antigens (reviewed in Moll and Berberich, 2001; Brandonisio, Spinelli and Pepe, 2004; Vanloubbeeck and Jones, 2004). Early experiments, pioneered by the group of Heidrun Moll, have shown that Langerhans cells pulsed with an L. major extract afforded protection from infection by skewing the immune responses towards Th1 (Flohe et al. 1998). Similarly, DCs loaded with a L. donovani soluble extract and expressing high levels of IL-12 induced protection in the mouse model of visceral leishmaniasis and had also a therapeutic effect (Ahuj et al. 1999). Moreover, co-administration of DCs with antimonial therapy resulted in complete clearance of parasites from liver and spleen, unlike DC immunisation alone which was not able to clear the infection from these organs (Ghosh et al. 2003). Further studies confirmed a crucial role for IL-12 in DC immunisation and demonstrated that loading DCs with defined antigens (LACK and gp63 amongst others) triggered protective Th1 responses against L. major (Berberich et al. 2003). However, DCs loaded with different gp63-derived peptides led to conflicting results ranging from significant protection to disease exacerbation depending on the peptide used (Tsagozis et al. 2004). Clearly, the choice of antigen is important since it can affect the immune response profile and determine host protection. Notwithstanding this, DC vaccination is a promising tool for inducing anti-leishmanial immunity, provided that the feasibility of large-scale delivery of such a vaccine can be addressed. In the meantime, if a widely accepted standard protocol for the production and preparation of DC-loaded vaccines can be developed, DC immunisation can become a useful tool for the preclinical evaluation of vaccine candidates.
ANTI-SANDFLY SALIVA COMPONENTS
Leishmania parasites are transmitted from one host to another during the sandfly bite as a suspension in sandfly saliva. Therefore, triggering immune responses against saliva components may indirectly enhance anti-leishmanial immunity. Early studies indicated that molecules in the sandfly saliva exacerbated the development of disease by facilitating the establishment of infection (Titus and Ribeiro, 1988; Kamhawi, 2000). On the other hand, prior exposure of mice to bites of uninfected sandflies seemed to confer protection from L. major infection. Protection was associated with a DTH response at the site of parasite injection (Kamhawi et al. 2000). Immunisation with molecules present in saliva, such as maxadilan (Morris et al. 2001) or a 15 kDa protein, SP15 (Valenzuela et al. 2001) also induced protection against cutaneous leishmaniasis.
The role of saliva molecules in the natural infection process is not understood; neither is the mechanism by which immune responses against them induce protection. Protective immunity can be directed towards immunomodulators in the saliva neutralising their ability to facilitate infection. Alternatively, the DTH response observed in all studies may modify the injection site environment and render it inhospitable for Leishmania (Belkaid et al. 2000). Anti-saliva vaccination opens an interesting avenue in Leishmania vaccinology, and it is likely that such a vaccine would have additive effects when administered together with an anti-Leishmania vaccine.
CROSS-IMMUNITY BETWEEN DIFFERENT LEISHMANIA SPECIES
One of the requirements of an ‘ideal’ anti-leishmanial vaccine is for it to be effective against more than one Leishmania species in order to protect individuals in areas where cutaneous and visceral leishmaniasis, for example, coexist. Although evidence of cross-protection was observed in humans who were refractory to L. mexicana infection following recovery from L. tropica-caused leishmaniasis (Adler and Gunders, 1964), such cross-protection is rare in humans (Mauel and Behin, 1982). A high degree of variability in cross-immunity between the New World Leishmania species has been observed in humans as well as in experiments in simian models (Lainson and Bray, 1966; Porrozzi, 2004; Lainson and Shaw, 1966, 1977). Lack of cross-protection was also observed in monkeys that recovered from L. donovani VL, but were susceptible to infection with L. panamensis (Lujan et al. 1990). In contrast, vervet monkeys exposed to L. donovani were resistant to L. major infection (Gicheru, Olobo and Anjili, 1997). Variable results were also obtained in cross-protection experiments in mice between L. major and L. mexicana, where a protection was influenced by the genetic background as well as gender of animals (Alexander and Phillips, 1978; Perez, Arredondo and Machado, 1979; Alexander, 1982, 1988). Other studies showed that L. major DHFR-TS knockouts were able to confer protection against L. amazonensis (Veras et al. 1999), and immunisation with heat-killed L. donovani promastigotes offered cross-protection against L. major challenge (Bebars et al. 2000).
The above studies highlight the complexity of the problem and the difficulties facing the design of a pan-Leishmania vaccine. Factors, such as virulence, genetic differences between Leishmania species as well as host genetic factors controlling the response to different Leishmania species (McMahon-Pratt et al. 2004) suggest that such a vaccine may not be feasible. However, the availability of the genome sequence of L. major, L. infantum and, in the future, other species may allow us to identify the genes responsible for the different disease phenotypes. This may lead to the identification of shared and species-specific antigens, which could be incorporated in a pan-Leishmania vaccine.
PROBLEMS CONCERNING THE LEISHMANIA VACCINE DEVELOPMENT
Vaccination is by far the most cost-effective means of control of infectious diseases. Several vaccines have proved very efficient in controlling infections, and some have even led to complete eradication of diseases such as smallpox, or almost complete eradication of polio (Paul, 2005). Nevertheless, there are many diseases for which no vaccines are available, and parasitic diseases fall into this category. That leishmaniasis ought to be controllable by vaccination seems indisputable in view of the body of evidence from studies in humans and animal models. Yet, no vaccine is currently on the market despite much effort. Therefore, the question arises what is the major problem in the anti-leishmanial vaccine development process?
Currently, there are more than 350 new candidate molecules for vaccines against 88 different pathogens under development at universities, research institutes and industry. Although many of these are directed against diseases of the developing world, there is a good chance that a large percentage may never make it into clinical use (Clemens and Jodar, 2005). Vaccines against diseases of the Third World such as leishmaniasis, malaria, schistosomiasis and several viral and bacterial infections are unappealing to the industry since the market is not sufficiently lucrative to recover the cost of development (300 to 800 million US dollars per vaccine) and make a profit (Plotkin, 2005). One solution is partial absorption of the vaccine costs by the developed world and subsidies by agencies distributing the vaccine in endemic regions. The World Health Organization and charitable foundations, such as the Bill and Melinda Gates Foundation, contribute greatly to the development of anti-parasitic vaccines and leishmaniasis is on their list. Public-private partnerships have also been suggested and the idea seems to have been taken on board by BigPharma.
Finances and politics aside, there are still unresolved scientific issues. One of the major problems facing a vaccine against cutaneous leishmaniasis for example, is the fact that despite causing cutaneous disease, the Old and New World parasites, L. major and L. mexicana/L. amazonensis, respectively, are markedly different. Phylogenetic analysis has revealed that L. major is as distant from L. mexicana/L. amazonensis as it is from L. donovani, which causes an entirely different disease (McMahon-Pratt et al. 2004). In inbred strains of mice the disease caused by the two species of Leishmania is different. C57BL/6 or C3H mice cure L. major infection, but they develop a chronic disease upon infection with the New World parasites (Alexander and Kaye, 1985). There are differences in virulence factors between these species as well as in the immune responses that they induce. For example, LPG is a virulence factor for L. major (Spath et al. 2000), but not for L. mexicana (Ilg, 2000). During L. major infection the protective role of Th1 responses has been established, at least in some experimental systems, but the role of IL-4 is still unclear. During L. mexicana infection IL-4 plays a major role in determining the severity of disease (McMahon-Pratt et al. 2004). Even more surprisingly, L. amazonensis is able to persist in the presence of Th1 responses, but it causes minimal disease in the complete absence of T cells (Soong et al. 1997). Altogether, these findings highlight major, but poorly understood differences in the immunobiology of parasites that seemingly cause the same disease. These may have implications for the vaccine development process since anti-CL vaccine may have different requirements for the Old World and New World leishmaniasis.
Selection of vaccine candidates has continued to be an extremely difficult problem. As outlined in this review, a plethora of antigens have been evaluated with mixed success depending on the formulation and the animal model used for testing. Complete protection has not been achieved so far and immunisation has usually led only to partial protection. In addition, opinions on the nature of the vaccine have been divided. Some argue that a vaccine against leishmaniasis should be molecularly defined, while others argue for a live attenuated vaccine.
The availability of the L. major and L. infantum genome sequences (Ivens et al. 2005), will undoubtedly lead to the identification of numerous molecularly defined antigens. If we are to follow this path, it is imperative to develop a coordinated approach to the evaluation of new candidates that minimises duplication of studies and maximises the use of resources.
If the live attenuated vaccine is considered, efficacy may need to be balanced by safety. Following natural infection parasites appear to persist probably for life (Solbach and Laskay, 2000); this persistence ensures protection from reinfection in cured individuals.
The process of vaccine validation has been hampered by the lack of unambiguous immunological or other correlates of protection. Early studies pointed to a correlation between DTH and protective immunity in humans and mice (De Rossell, Bray and Alexander, 1987), but several conflicting reports suggest that caution should be exercised before any extrapolation from mice to humans can be made.
Animal models of cutaneous leishmaniasis indicate that Th1 responses are essential for protection by vaccination. This has been usually predicated on the induction of high levels of IFN-γ and low levels of IL-4. However, recent studies indicate that even vaccines triggering high levels of IFN-γ do not protect in the presence of high levels of the regulatory cytokine IL-10 (Stober et al. 2005). Moreover, IL-4 and IL-10 act together in the presence of exacerbatory antigens (Roberts et al. 2005). Hence, it would appear that IL-10 may be the most appropriate cytokine to serve as an indicator of failure or success of vaccination.
To complicate things, the use of the mouse model for disease mimics only some aspects of the human disease. For example, the outcome of infection and the immune response induced is affected by the strain of mouse used, by the site of infection (Baldwin et al. 2003) and type of a challenge, i.e. by sandfly bite or injection by syringe.
The immune responses needed for protection in humans are not surprisingly less clear than in the mouse models. For currently available vaccines such as hepatitis B, influenza, measles or diphtheria, where T cell responses are induced, their contribution to protective immunity is not well understood. Bacille Clamette-Guerin (BCG) is the only vaccine in use that relies on the generation of T cell responses, thus can be used as a prototypic vaccine for other diseases, leishmaniasis in particular (Lambert, Liu and Siegrist, 2005). Based on BCG, a Leishmania vaccine needs to trigger strong IFN-γ production through Th1 responses as well as activation of CD8+ T cells in order to develop and maintain the protective response. Another, essential requirement seems to be IL-12 that is a critical cytokine in the initiation and maintenance of immunity, as well as a very effective adjuvant (Scott et al. 2004).
The essential requirement for any vaccine is long-lasting immunity. Recent insights into the generation of immunological memory (Wong et al. 2004; Badovinac et al. 2005) indicate that central memory T cells are generated during the early stages of infection and persist in the absence of antigen (Zaph et al. 2004). Thus, defining the requirements for the generation and maintenance of central memory T cells is crucial for Leishmania vaccine development. If long-lived central memory T cells do not require parasite persistence, it should be possible for sub-unit vaccine formulations to direct the immune response towards central memory formation. However, this may not be suitable for a therapeutic vaccine, which would require a strong and rapid effector T cell expansion upon delivery. The understanding of memory formation and maintenance is progressing rapidly, but it may be some time before these concepts can be translated into vaccine design.
CONCLUSIONS
Preventive vaccines are recognised as the best and most cost-effective protection measure against pathogens, and Leishmania is no exception. Leishmania vaccine development has proven to be a difficult and challenging task, which is mostly hampered by inadequate knowledge of parasite pathogenesis and the complexity of immune responses needed for protection. Nevertheless, one of the candidates, Leish-111f has reached Phase I clinical testing following promising results in animal model studies. Several new antigens are being assessed and the completion of L. major and L. infantum genomes will, no doubt, unveil more promising candidates. Our understanding of immune responses and memory formation following immunisation, while still fragmentary, offers hope for development of new strategies allowing for effective T cell vaccines. The main concerns are reliable correlates of immunity that need to be developed in order to evaluate vaccines, as well as a need for a uniform testing system for new vaccine candidates. Then, the issues of delivery systems, antigen formulation and adjuvant would have to be resolved. There is a pressing need to develop better animal models for visceral leishmaniasis that can help the design of a vaccine to control both canine and human disease in endemic areas. Currently, there seem to be as many problems and questions as there are solutions, but given the rapid progress in the vaccinology field, a successful anti-Leishmania vaccine should be achievable.
ACKNOWLEDGMENTS
The authors would like to thank Dr Katherine Kedzierska for critical review of the manuscript. The authors are supported by the Australian National Health and Medical Research Council and the NHMRC/ARC Network for Parasitology.



