Introduction
World-wide movement and commercialization of live animals can lead to the introduction and establishment of infectious disease into disease-free areas (Peeler et al., Reference Peeler, Oidtmann, Midtlyng, Miossec and Gozlan2011; Lymbery et al., Reference Lymbery, Morine, Kanani, Beatty and Morgan2014). Examples are the squirrel poxvirus, causing the decline of the native red squirrel in the UK and linked to the introduction of the grey squirrel from North America (Rushton et al., Reference Rushton, Lurz, Gurnell, Nettleton, Bruemmer, Shirley and Sainsbury2006) and the swim bladder nematode Anguillicola crassus, causing the decline of the European eel and linked to the introduction of the Japanese eel (Lymbery et al., Reference Lymbery, Morine, Kanani, Beatty and Morgan2014). The decline of European crayfish species is linked to the introduction of North American crayfish, which can carry the parasite Aphanomyces astaci in melanised cuticle as a benign infection (Unestam and Weiss, Reference Unestam and Weiss1970; Alderman et al., Reference Alderman, Polglase, Frayling and Hogger1984; Diéguez-Uribeondo and Söderhäll, Reference Diéguez-Uribeondo and Söderhäll1993; Holdich et al., Reference Holdich, Reynolds, Souty-Grosset and Sibley2009; Jussila et al., Reference Jussila, Vrezec, Makkonen, Kortet, Kokko and Canning-Clode2015). The likely first introduction of A. astaci was in the mid-19th century in Italy, with the first recorded European crayfish (Astacus astacus – noble crayfish) mass mortalities described by Martinati (Reference Martinati1862) and Ninni (Reference Ninni1865). Due to consecutive independent introductions of North American crayfish species, the parasite rapidly spread across Europe reaching the Balkan Peninsula, the Black Sea, Turkey and Russia in the east, spreading across Western Europe, reaching Spain in the south, and entering the British Isles and Northern Europe (Unestam, Reference Unestam1972; Alderman et al., Reference Alderman, Polglase, Frayling and Hogger1984; Alderman, Reference Alderman1996; Jussila et al., Reference Jussila, Vrezec, Makkonen, Kortet, Kokko and Canning-Clode2015).
The identification of A. astaci is currently based on molecular diagnostic tests, which can reliably detect the pathogen from a wide range of substrates, including moribund crayfish retrieved during crayfish plague outbreaks (Oidtmann et al., Reference Oidtmann, Geiger, Steinbauer, Culas and Hoffmann2006), asymptomatic (carrier) crayfish (Vrålstad et al., Reference Vrålstad, Knutsen, Tengs and Holst-Jensen2009) and environmental samples (Strand et al., Reference Strand, Holst-Jensen, Viljugrein, Edvardsen, Klaveness, Jussila and Vrålstad2011). To date, five genotype groups (A, B, C, D and E) have been genetically characterized from pure cultures by random amplified polymorphic DNA polymerase chain reaction (RAPD-PCR) (Huang et al., Reference Huang, Cerenius and Söderhäll1994; Diéguez-Uribeondo et al., Reference Diéguez-Uribeondo, Huang, Cerenius and Söderhäll1995; Kozubíková et al., Reference Kozubíková, Viljamaa-Dirks, Heinikainen and Petrusek2011). Genotype group A is thought to be related to the first introduction of A. astaci into Europe, having been isolated from crayfish plague outbreaks in noble crayfish (Astacus astacus) and narrow-clawed crayfish (Astacus leptodactylus) (Huang et al., Reference Huang, Cerenius and Söderhäll1994). Genotype group B has been introduced into Europe and isolated from signal crayfish (Pacifastacus leniusculus) and from European freshwater crayfish (Huang et al., Reference Huang, Cerenius and Söderhäll1994; Lilley et al., Reference Lilley, Cerenius and Söderhäll1997). Genotype group C has been found in a single signal crayfish imported from Canada into Sweden (Huang et al., Reference Huang, Cerenius and Söderhäll1994). Genotype group D has been isolated from red swamp crayfish (Procambarus clarkii) (Diéguez-Uribeondo et al., Reference Diéguez-Uribeondo, Huang, Cerenius and Söderhäll1995; Rezinciuc et al., Reference Rezinciuc, Galindo, Montserrat and Diéguez-Uribeondo2014) and genotype group E has been isolated from spinycheek crayfish (Orconectes limosus) (Kozubíková et al., Reference Kozubíková, Viljamaa-Dirks, Heinikainen and Petrusek2011).
Beside the RAPD-PCR, amplified fragment length polymorphism analysis has been developed to genotype A. astaci from pure cultures (Rezinciuc et al., Reference Rezinciuc, Galindo, Montserrat and Diéguez-Uribeondo2014). However, axenic cultures can be challenging to establish (Oidtmann et al., Reference Oidtmann, Schmid, Rogers and Hoffmann1999).
Advances in molecular biology methods have led to the development of new genotyping tools, which can be applied directly to crayfish samples instead of pure cultures and that allow for re-interrogation of historic outbreak samples (Vrålstad et al., Reference Vrålstad, Strand, Grandjean, Kvellestad, Hastein, Knutsen, Taugbøl and Skaar2014). Examples of these techniques that can distinguish all five currently known genotype groups (A, B, C, D and E) and that can be applied directly to infected crayfish cuticle are based on microsatellites (Grandjean et al., Reference Grandjean, Vrålstad, Diéguez-Uribeondo, Jelić, Mangombi, Delaunay, Filipová, Rezinciuc, Kozubíková-Balcarová, Guyonnet, Viljamaa-Dirks and Petrusek2014) or genotype-specific regions (Minardi et al., Reference Minardi, Studholme, van der Giezen, Pretto and Oidtmann2018). However, both techniques require high A. astaci levels and cannot be applied to characterize crayfish plague outbreaks samples with low levels of infection or to samples from carriers (North American crayfish), which can harbour the parasite in melanised cuticle as a benign infection (Vrålstad et al., Reference Vrålstad, Strand, Grandjean, Kvellestad, Hastein, Knutsen, Taugbøl and Skaar2014; Makkonen et al., Reference Makkonen, Jussila, Panteleit, Keller, Schrimpf, Theissinger, Kortet, Martín-Torrijos, Sandoval-Sierra, Diéguez-Uribeondo and Kokko2018). The sensitivity of these techniques is low because they target single-copy nuclear DNA sequences (Makkonen et al., Reference Makkonen, Jussila, Panteleit, Keller, Schrimpf, Theissinger, Kortet, Martín-Torrijos, Sandoval-Sierra, Diéguez-Uribeondo and Kokko2018). As eukaryotic cells generally contain many mitochondria and therefore many copies of mitochondrial DNA (mtDNA) (Waugh, Reference Waugh2007; Luo et al., Reference Luo, Zhang, Ho, Xu, Zhang, Shi, Cameron and Zhu2011), the sensitivity of a genotyping tool may be improved by targeting mtDNA. Recently, a more sensitive haplotyping technique based on A. astaci mtDNA has been developed (Makkonen et al., Reference Makkonen, Jussila, Panteleit, Keller, Schrimpf, Theissinger, Kortet, Martín-Torrijos, Sandoval-Sierra, Diéguez-Uribeondo and Kokko2018). However, while this technique can distinguish new uncharacterized haplotypes, it is not able to distinguish all known genotype groups (i.e. genotype groups A and C cannot be mutually distinguished, and both groups gather together in the same haplogroup) (Makkonen et al., Reference Makkonen, Jussila, Panteleit, Keller, Schrimpf, Theissinger, Kortet, Martín-Torrijos, Sandoval-Sierra, Diéguez-Uribeondo and Kokko2018).
The genotyping techniques developed to discriminate A. astaci genotypes have been used in numerous epidemiological and phylogenetic studies allowing a better understanding of the history and spread of this disease in Europe or in specific geographic regions (such as Norway and Finland) and the evolutionary relationship of A. astaci genotypes with its hosts (Lilley et al., Reference Lilley, Cerenius and Söderhäll1997; Viljamaa-Dirks et al., Reference Viljamaa-Dirks, Heinikainen, Torssonen, Pursiainen, Mattila and Pelkonen2013; Rezinciuc et al., Reference Rezinciuc, Galindo, Montserrat and Diéguez-Uribeondo2014; Vrålstad et al., Reference Vrålstad, Strand, Grandjean, Kvellestad, Hastein, Knutsen, Taugbøl and Skaar2014; Kaldre et al., Reference Kaldre, Paaver, Hurt and Grandjean2017; Kokko et al., Reference Kokko, Harlioglu, Aydin, Makkonen, Gökmen, Aksu and Jussila2018; Makkonen et al., Reference Makkonen, Jussila, Panteleit, Keller, Schrimpf, Theissinger, Kortet, Martín-Torrijos, Sandoval-Sierra, Diéguez-Uribeondo and Kokko2018; Panteleit et al., Reference Panteleit, Keller, Diéguez-Uribeondo, Makkonen, Martín-Torrijos, Patrulea, Pîrvu, Preda, Schrimpf and Pârvulescu2018). Being able to reliably identify A. astaci genotypes is an important tool to understand the crayfish plague agent.
In the current study, we exploit genotype-specific single nucleotide variants (SNVs) present in the mtDNA of each known A. astaci genotype groups (A, B, C, D and E). SNVs were bioinformatically identified to develop a sensitive genotyping technique based on PCR amplification, and subsequent restriction digestion (genotypes A, C, D, and E) or semi-nested PCR (genotype B). The genotyping assays were validated using pure A. astaci cultures, other Aphanomyces species, oomycetes and fungi, historical samples available in the Centre for Environment, Fisheries and Aquaculture Science (Cefas) laboratory (including UK outbreaks and carriers samples) and samples from a recent Italian outbreak of crayfish plague.
Materials and methods
Aphanomyces astaci mitochondrial genome assembly and bioinformatic identification of genotype-specific SNVs in restriction sites
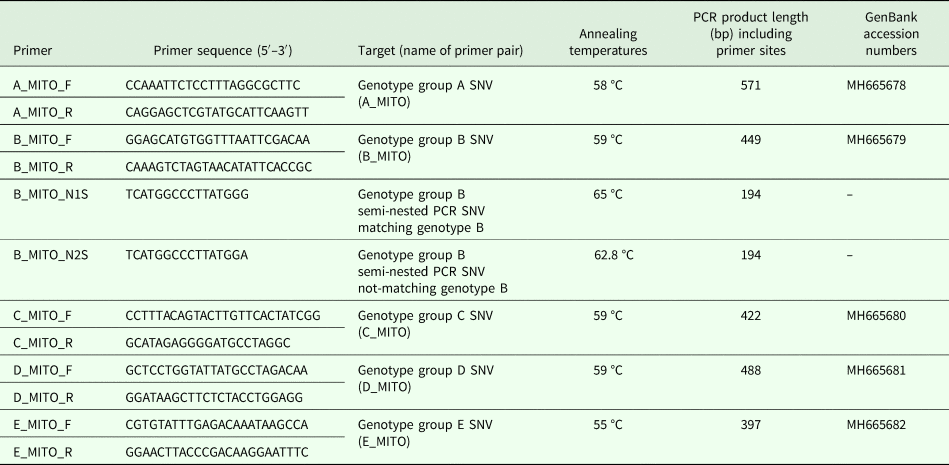
Isolate genotypes were defined as per Cefas Oomycete Culture Collection (OCC) record system, based on RAPD-PCR analysis (Huang et al., Reference Huang, Cerenius and Söderhäll1994) and nomenclature of genotype groups followed Svoboda et al. (Reference Svoboda, Mrugała, Kozubíková-Balcarová and Petrusek2017). Genomic data from 11 A. astaci isolates belonging to the five known genotype groups (Supplementary Table S1) and generated earlier (Minardi et al., Reference Minardi, Studholme, van der Giezen, Pretto and Oidtmann2018) were used to assemble an A. astaci reference mitochondrial genome and to retrieve genotype-specific SNVs as follows. Using BWA (Li, Reference Li2013) and SAMtools (Li et al., Reference Li, Handsaker, Wysoker, Fennell, Ruan, Homer, Marth, Abecasis and Durbin2009), trimmed pair-end reads of A. astaci isolate D2 (genotype B – used as reference mitochondrial genome) were aligned against a related oomycete mitochondrion genome retrieved from NCBI (Saprolegnia ferax, GenBank accession number: AY534144.1). Mapped reads were extracted using samtools view from the SAMtools package and assembled using SPAdes (Bankevich et al., Reference Bankevich, Nurk, Antipov, Gurevich, Dvorkin, Kulikov, Lesin, Nikolenko, Pham, Prjibelski, Pyshkin, Sirotkin, Vyahhi, Tesler, Alekseyev and Pevzner2012). To identify genotype group-specific SNVs, paired-end reads from the 11 A. astaci isolates were aligned against the reference mitochondrial genome using BWA and SAMtools. For each genotype group, genotype group-specific SNVs were identified using a previously described method (Mazzaglia et al., Reference Mazzaglia, Studholme, Taratufolo, Cai, Almeida, Goodman, Guttman, Vinatzer and Balestra2012; Clarke et al., Reference Clarke, Studholme, Hayes, Runde, Weisberg, Cai, Wroblewski, Daunay, Wicker, Castillo and Vinatzer2015) and those that fell within a restriction site (and thus leading to a restriction polymorphism) were identified. Flanking sequences and SNVs were inspected in IGV (Thorvaldsdóttir et al., Reference Thorvaldsdóttir, Robinson and Mesirov2013), before designing primers to amplify regions of about 500 bp of length (Table 1).
Table 1. Primers developed in this study to amplify Aphanomyces astaci regions containing genotype group-specific SNVs, PCR specific annealing temperatures and expected PCR product length

Primers (synthesized by Eurofins Genomics) were tested for secondary structures formation using the program Oligo Analysis Tool (Eurofins Genomics). Hyphen (–) = sequence not submitted as amplifying half portion of sequence MH665679.
PCR protocols and reactions, primers sensitivity
PCR reactions contained 1x GoTaq® Hot Start Green Master Mix, 10 mm of forward primer, 10 mm of reverse primer, 78.3–380 ng of DNA template (pure cultures) and nuclease-free water to 25 µL, final volume. Amplification of the targeted sequences was conducted on a Bio-Rad T100 thermal cycler with the following conditions: 95 °C for 2 min; 35 cycles of 95 °C for 1 min, annealing (for temperatures see Table 1) for 45 s, 72 °C for 45 s; 72 °C for 5 min. Semi-nested PCR (primer pair: B_MITO_N1S and B_MITO_R; B_MITO_N2S and B_MITO_R) conditions: 95 °C for 2 min; 20 cycles of 95 °C for 1 min, annealing (for temperature see Table 1) for 45 s, 72 °C for 30 s; 72 °C for 5 min. For each PCR reaction, negative controls without DNA template and positive controls were included. Prior digestion or semi-nested PCR, amplicons were checked on 1% agarose gel stained with Midori Green Advance. Positive PCR products were purified with GeneJET PCR Purification Kit (Thermo Fisher Scientific), sequenced in Eurofins MWG Operon commercial sequencing facility and sequences visualized with BioEdit version 7.0.8 (Hall, Reference Hall1999). Primer pairs sensitivity was tested on A. astaci pure culture genomic DNA (gDNA) dilution series (starting concentrations: A. astaci Da 78.3 ng µL−1; A. astaci D2 314 ng µL−1; A. astaci KV 289 ng µL−1; A. astaci Pc 380 ng µL−1; A. astaci KB13 176 ng µL−1). Tested dilution series of each A. astaci gDNA stretched to 10−9 ng µL−1, corresponding to agent level (A) < A1 as defined by Vrålstad et al. (Reference Vrålstad, Knutsen, Tengs and Holst-Jensen2009).
Enzymatic digestion
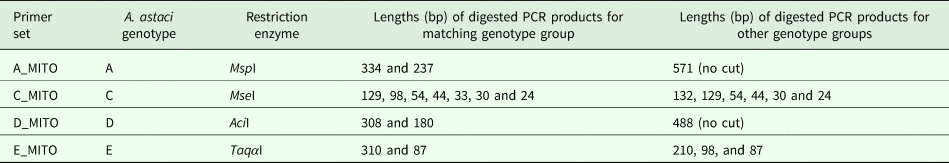
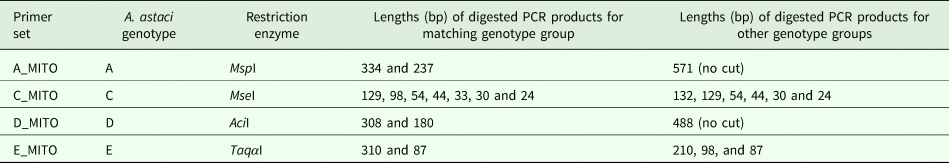
After amplification of the regions containing the genotype group-specific SNVs, enzymatic digestion was applied to PCR products. The restriction enzymes (New England Biolabs) bioinformatically identified to be associated with restriction sites containing the SNVs were: MspI (A_MITO); Msel (C_MITO); Acil (D_MITO); TaqαI (E_MITO) (Table 2). Each digestion contained 300 ng of non-purified PCR product and was mixed with 2 µL of 10x CutSmart Buffer (New England Biolabs), 0.3 µL of restriction enzyme and nuclease-free water up to 20 µL. The mixture was incubated following manufacturer instructions at 37 °C for 1 h (AciI, MseI and MspI) or at 65 °C for 1 h (TaqαI). Digested products were checked by gel electrophoresis using 2% agarose gel stained with Midori Green Advance stain.
Table 2. Restriction enzymes and expected lengths of the digested PCR product for each genotype. Enzymes from New England Biolabs.
Genotyping A. astaci isolates and other species tested for primer specificity
Primer and enzymatic digestion specificity were tested on gDNA from A. astaci isolates (Supplementary Table S1) and other oomycete and fungal isolates available in the Cefas OCC (Achlya racemosa; A. frigidophilus isolates AP5, RP1, and RP2; A. invadans isolates GWR, NJM0002, NJM8997, NJM9030, NJM9701; A. invadans-like NJM9510; A. laevis 7093; Leptolegnia caudata; Phoma-like; Pythium monospermum; P. flevoense; Saprolegnia furcata 1–12; S. parasitica RP5). A total of 17 non-A. astaci species previously extracted as described in Minardi et al. (Reference Minardi, Studholme, van der Giezen, Pretto and Oidtmann2018) were used.
Outbreaks and carrier samples
The new genotyping assays were tested on DNA extracts from white clawed crayfish (Austropotamobius pallipes) and from A. astaci pure cultures (Supplementary Table S2) isolated during a crayfish plague outbreak in Italy, previously genotyped by RAPD-PCR and microsatellites (Pretto et al., Reference Pretto, Tosi, Sandoval-Sierra, Grandjean, Manfrin and Diéguez-Uribeondo2014), and sequencing of single-copy genotype-specific regions (Minardi et al., Reference Minardi, Studholme, van der Giezen, Pretto and Oidtmann2018) and archived in the Istituto Zooprofilattico Sperimentale delle Venezie (IZSVe). Moreover, DNA extractions archived in Cefas from infected white-clawed crayfish (A. pallipes) cuticles collected from UK sites of outbreak events during 2007–2014 (Supplementary Table S2) and DNA extractions archived in Cefas from North American carrier crayfish P. leniusculus (signal crayfish) cuticles collected from various locations in watercourses in England and Wales during 2009 and 2010 (Supplementary Table S3) were tested with the new genotyping assays. This last set of samples was previously screened for the prevalence of A. astaci in the UK by James et al. (Reference James, Nutbeam-Tuffs, Cable, Mrugala, Viñuela-Rodriguez, Petrusek and Oidtmann2017). PCR cycle numbers for Cefas archived samples was increased from 35 to 45 cycles. Before genotyping, total DNA was extracted from both types of samples at Cefas immediately after sampling by a BioRobot® EZ1 workstation and EZ1 DNA Tissue kit (Qiagen) following manufacturer protocol. The presence of A. astaci was tested by PCR amplification with the internal transcribed spacer (ITS) specific primers ITS1 and ITS4 and 42–640 primer pairs and PCR products amplification (OIE, 2018) for outbreak samples, or by qPCR (Vrålstad et al., Reference Vrålstad, Knutsen, Tengs and Holst-Jensen2009) for carrier samples.
Results
Bioinformatic identification of genotype group-specific SNVs
Genomic sequence reads are available from the Sequence Read Archive (Leinonen et al., Reference Leinonen, Sugawara and Shumway2011) under study accession number SRP168228. The genome sequencing data will be described in full in a separate manuscript currently in preparation.
The number of genotype group-specific SNVs retrieved bioinformatically in the mitochondrial genome of each A. astaci genotypes corresponded to 6 for genotype group A, 4 each for genotype groups B and E, 2 for genotype group C and 92 for genotype group D. For genotype groups A, C, D and E, it was possible to identify at least one SNV in a restriction site and the SNVs chosen for developing the genotyping assays were: genotype group A, restriction site ‘CCGG’, digested by restriction enzyme MspI; genotype group C, restriction site ‘TTAA’, digested by restriction enzyme MseI; genotype group D, the restriction site ‘CCGC’, digested by restriction enzyme AciI; genotype group E, restriction site ‘TCGA’, digested by the enzyme TaqαI (Table 2). For genotype group B, as no SNVs were in a restriction site, a semi-nested PCR protocol based on the presence/absence of PCR product in the nested PCR was developed to exploit one of the SNVs detected. One set of outer primers (Table 1, primer pair B_MITO) was designed for the first round PCR and two primers of equal size and nucleotide sequence, except for the last nucleotide, were designed for the semi-nested PCR. Amplifying the PCR product obtained from the first round with the semi-nested primers, the presence or absence of the second band of smaller size in comparison to the original PCR product on the agarose gel indicates that the primers matched the variant in the template (Chen and Sullivan, Reference Chen and Sullivan2003). By using one nested primer at a time in the PCR reactions [B_MITO_N1S, semi-nested PCR reaction control primer matching genotype group B (RcB) and B_MITO_R; B_MITO_N2S, semi-nested PCR reaction control primer non-matching genotype B group (RcA) and B_MITO_R], the PCR product can be ascribed to genotype group B or not.
Evaluation of primer sensitivity, primers specificity and genotyping A. astaci pure cultures
Initial evaluation of primer pair sensitivity was tested on diluted A. astaci gDNA and primers were able to detect between 78.3 × 10−7 ng µL−1 and 38 × 10−6 ng µL−1 of A. astaci gDNA, corresponding to 0.2 and 1.2 PCR forming units respectively, and equal to agent level A1 (and below) estimated following Vrålstad et al. (Reference Vrålstad, Knutsen, Tengs and Holst-Jensen2009).
The primer sets (A_MITO, B_MITO, C_MITO and D_MITO) successfully amplified the mtDNA targeted sequences from A. astaci isolates and the other oomycetes tested (results not shown). Primer pair E_MITO amplified DNA regions only in all A. astaci, A. frigidophilus, A. invadans NJM9701 and GWR, and the oomycetes S. furcata and A. laevis (results not shown).
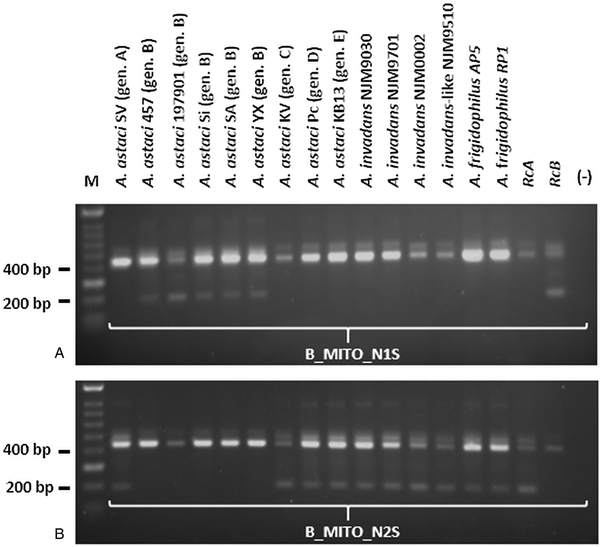
PCR products obtained by amplification with A_MITO, C_MITO, D_MITO and E_MITO primer pairs were subsequently cut with their associated restriction enzymes, while the PCR products obtained by amplification with B_MITO primer pairs were used in the semi-nested PCR approach. For genotype group A, the enzymatic digestion occurred only for the PCR product corresponding to A. astaci SV and Da (isolates both belonging to genotype group A), while it did not occur for the other A. astaci genotypes and oomycetes tested (Fig. 1A and Supplementary Fig. S1A). For genotype group C, the enzymatic digestion occurred for all PCR fragments, however the pattern of the bands on the gels differ from the ones produced by A. astaci KV (isolate belonging to genotype group C) (Fig. 1B and Supplementary Fig. S1B). For genotype group D, the enzymatic digestion did not occur for most isolates tested, and while A. invadans-like NJM9510 and S. furcata presented a different band pattern in comparison to A. astaci Pc (isolate belonging to genotype group D), isolates L. caudata and Phoma-like showed a similar pattern to the positive control (Fig. 1C and Supplementary Fig. S1C). Nonetheless, sequences of the PCR products of isolates L. caudata and Phoma-like revealed the presence of the SNV in the restriction site for A. astaci Pc (belonging to genotype group D) thus the same band pattern but the overall sequences differed between the isolates (result not shown). For genotype group E, the enzymatic digestion occurred for all PCR fragments, however the pattern of the bands on the gels differed from the ones produced by A. astaci KB13 (belonging to genotype group E) (Fig. 1D and Supplementary Fig. S1D). PCR products obtained by amplification with B_MITO primer set were submitted to semi-nested PCR. Amplification with B_MITO_N1S primer (primer with SNV matching genotype group B) occurred only from the isolates belonging to genotype group B (A. astaci D2, 197901, 457, Sa, Si, YX) and no amplification occurred from A. astaci isolates belonging to other genotype groups nor from the other oomycetes tested (Fig. 2A and Supplementary Fig. S2A). Conversely, no amplification occurred from genotype group B isolates using the primer B_MITO_N2S (primer with SNV non-matching genotype B), while it occurred from A. astaci belonging to the other genotype groups, A. invadans, A. invadans-like, A. frigidophilus and the oomycete A. laevis (Fig. 2B and Supplementary Fig. S2B).

Fig. 1. Enzymatic digestions of PCR products obtained from Aphanomyces spp. isolates amplified with the mitochondrial primers developed in the present study. A: A_MITO; B: C_MITO; C: D_MITO; D: E_MITO. M = Bioline HyperLadder II. Gen. = genotype.
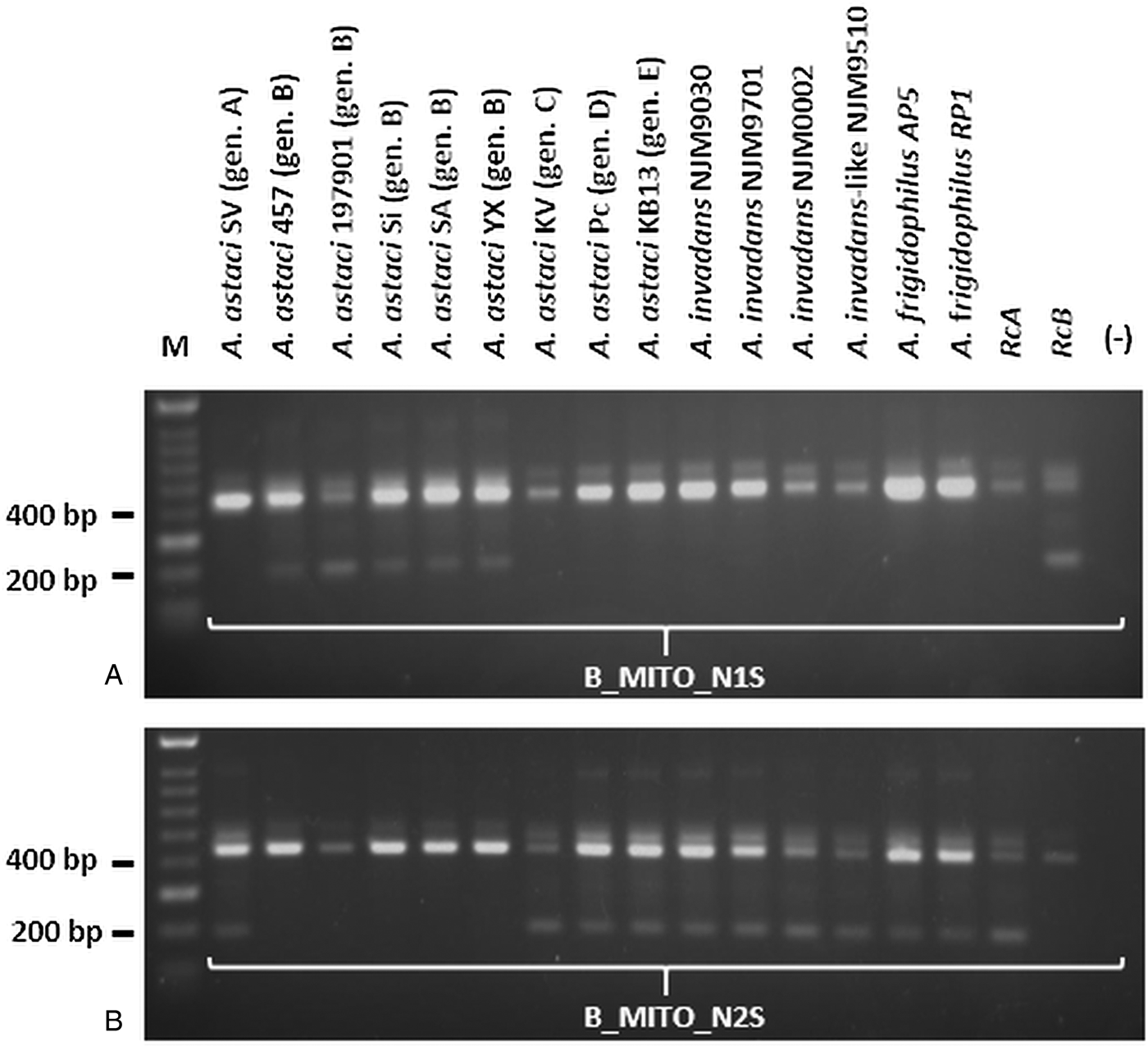
Fig. 2. Semi-nested PCR results of Aphanomyces spp. isolates amplified with the mitochondrial primers developed in the present study. A: B_MITO_N1S; B: B_MITO_N2S. M = Bioline HyperLadder II. Gen. = genotype. RcA = PCR reaction control (A. astaci Da genotype A). RcB = PCR reaction control (A. astaci D2 genotype B). (-) = PCR negative control.
Genotyping the Italian outbreak and Cefas archive samples
The genotyping assays were tested on the 2014 Italian outbreak samples. Amplification products were obtained with all primers sets (results not shown). The restriction digestions assays for genotype groups C, D and E were negative (i.e. not matching the positive controls) indicating the absence of A. astaci belonging to these genotype groups (Supplementary Fig. S3A and S3B). Likewise, the semi-nested PCR assay using primers B_MITO_N1S and B_MITO_N2S on the first-round PCR products was negative, indicating the absence of this genotype group in the samples (Supplementary Fig. S3C). On the other hand, the restriction digestions of all the PCR products amplified with A_MITO primer pair matched the positive control (Supplementary Fig. S3A), results confirmed by sequencing the PCR products, which matched the A. astaci genotype group A mtDNA genomic sequence.
The restriction digestion assays and the semi-nested PCR were tested on Cefas’ archive samples (outbreaks and carriers samples). Amplification of PCR products was achieved with all primer sets in most of the samples tested (results not shown). PCR products obtained with primer pairs A_MITO, C_ MITO, D_ MITO and E_ MITO were subjected to restriction digestion assays, and PCR products obtained with primers pairs B_MITO were subjected to semi-nested PCRs. While all outbreak samples were negative for the presence of A. astaci genotype groups C, D and E, the majority were positive for the presence of A. astaci genotype group B and outbreak PM21018 sample number 2 was positive for A. astaci genotype group A (Supplementary Table S4). The genotyping assay detected the simultaneous presence of genotype groups A and B in outbreak PM21018. All carrier samples tested in this study were negative for the presence of genotype groups A, C, D and E and 12 out of 16 samples tested resulted positive for the presence of A. astaci genotype group B (Supplementary Table S5). Three samples were positive at both B_MITO semi-nested primers, therefore first round PCR products were analysed to confirm sequence identity (Supplementary Fig. S4). For sample III-28-C, the SNV did not match the one from A. astaci mitochondrial reference sequence for genotype group B, and the overall sequence of the PCR product presented some differences (Supplementary Fig. S4). The SNV of sample XVII-9a2-C did not match the one from the reference sequence, but the PCR product shared similar sequence (Supplementary Fig. S4). The SNV of sample XVIII-16a1-C matched the mitochondrial reference SNV, and the PCR product presented a highly similar sequence to the reference sequence (Supplementary Fig. S4).
Discussion
In the present study, we developed and tested new genotyping assays based on mtDNA that allow the identification of all currently known A. astaci genotype groups from pure cultures, historical outbreaks and carrier samples. The genotyping assays are a combination of four restriction digestion assays and a semi-nested PCR. For genotype groups A, C, D and E it was possible to detect SNVs within restriction sites, the SNVs unique to genotype group B on the mtDNA were not in restriction sites and therefore a semi-nested PCR was developed. The capability of these bioinformatically developed genotyping assays was validated on A. astaci pure cultures, and its specificity tested on 17 non-A. astaci isolates. The assays designed in silico worked in vitro on pure cultures of A. astaci and it was possible to confirm the genotype groups of the pure cultures (Figs 1 and 2). The primer pairs did not specifically amplify A. astaci mtDNA only, as amplification was obtained from the other species tested (results not shown). However, when applying the restriction digestion (for genotype groups A, C, D and E), the semi-nested PCR assay (for genotype group B) and subsequent sequencing of PCR products (for genotype group D), the assays were A. astaci and genotype group-specific. The PCR products from non-A. astaci species were either not digested (genotype groups A and D restriction digestion), or the pattern of the bands on the gels differed from the ones produced by the positive control (genotype groups C, D and E restriction digestions), or PCR products were not amplified by the semi-nested primers (Figs 1 and 2, and Supplementary Fig. S1). For genotype group D, L. caudata and Phoma-like isolates showed a similar pattern to the positive control, but overall sequence difference between the isolates. While Leptolegnia caudata is a widely distributed aquatic phytosaprobiont oomycete (Czeczuga et al., Reference Czeczuga, Godlewska, Czeczugasrmeniuk, Semeniuk and Muszyńska2015), the Phoma genus (Ascomycota) includes a wide group of ubiquitous phytopathogenetic organisms, which colonize soil, organic matter, plants and water sources (Bennett et al., Reference Bennett, Ponder and Garcia-Diaz2018). Due to their ubiquitous presence in water and their ability to opportunistically parasitize aquatic animals and colonize dead animals as necrotrophs (Czeczuga et al., Reference Czeczuga, Kiziewicz and Gruszka2004; Bennett et al., Reference Bennett, Ponder and Garcia-Diaz2018), cross-reactions of the assays developed in this study with these species and other untested oomycetes or fungi cannot be excluded. Nonetheless, the possibility to sequence and compare the PCR products increases the confidence in the assay results, the approach followed here to separate L. caudata and Phoma species from A. astaci.
The genotyping assays were then tested on a crayfish plague outbreak in Italy and Cefas archive samples. For the Italian outbreak, the genotyping assays matched the result of genotyping techniques previously applied to the same set of samples [genotype A, identified by RAPD-PCR and microsatellites by Pretto et al. (Reference Pretto, Tosi, Sandoval-Sierra, Grandjean, Manfrin and Diéguez-Uribeondo2014), and single-copy region analysis by Minardi et al. (Reference Minardi, Studholme, van der Giezen, Pretto and Oidtmann2018)]. While PCR products were obtained from all the Italian samples with all primer sets, it was not possible to obtain PCR products from all Cefas archive (either outbreaks or carriers). Cefas archive samples were determined to be positive for A. astaci at the time of sampling by PCR and sequencing (outbreaks samples, primers 42 and 640, unpublished data) and by qPCR (carrier samples, Tuffs and Oidtmann unpublished data). The failure to amplify a PCR product with the new genotyping assays does not exclude the absence of A. astaci or novel genotypes and genotype groups in those samples. However, the presence of contaminants, inhibitors, degraded DNA, or other oomycetes could explain the failure of amplification from those samples (Wilson, Reference Wilson1997). The assays developed in the present study are based on the typical genotype groups defined by RAPD-PCR and cannot confirm the presence of a novel A. astaci genotype and/or genotype group. As variations within the known A. astaci genotype groups by means of unusual microsatellites patterns have been reported from various geographical regions (Grandjean et al., Reference Grandjean, Vrålstad, Diéguez-Uribeondo, Jelić, Mangombi, Delaunay, Filipová, Rezinciuc, Kozubíková-Balcarová, Guyonnet, Viljamaa-Dirks and Petrusek2014; Kozubíková-Balcarová et al., Reference Kozubíková-Balcarová, Beran, Ďuriš, Fischer, Horká, Svobodová and Petrusek2014; James et al., Reference James, Nutbeam-Tuffs, Cable, Mrugala, Viñuela-Rodriguez, Petrusek and Oidtmann2017; Mrugala et al., Reference Mrugala, Kawai, Kozubíková-Balcarová and Petrusek2017), the presence of novel genotypes and genotype groups not yet characterized by RAPD-PCR cannot be excluded from the carrier samples.
All UK outbreak samples from which a PCR product was obtained were positive for the presence of A. astaci genotype group B and negative for genotype groups C, D and E. Alongside genotype group B, outbreak PM21018 (River Wissey) was also positive for A. astaci genotype group A. All carrier samples from which a PCR product was obtained were unambiguously positive for A. astaci genotype group B, except of carrier samples III-28-C, XVII-9a2-C and XVIII-16a1-C, which presented an unusual semi-nested PCR profile (amplification achieved with both semi-nested primers). Sequenced PCR products from these samples suggest that A. astaci genotype group B is present but in concomitance with another unknown A. astaci genotype or genotype group.
In Great Britain, crayfish plague outbreaks have been linked to the introduction of infected signal crayfish from Sweden (Holdich and Reeve, Reference Holdich and Reeve1991; Lilley et al., Reference Lilley, Cerenius and Söderhäll1997; Holdich et al., Reference Holdich, Reynolds, Souty-Grosset and Sibley2009, Reference Holdich, James, Jackson and Peay2014), with crayfish plague-related mortalities first detected in the summer of 1981 (Alderman et al., Reference Alderman, Polglase, Frayling and Hogger1984; Polglase and Alderman, Reference Polglase and Alderman1984; Alderman, Reference Alderman2003). RAPD-PCR genotyping of A. astaci pure cultures isolated in Sweden before 1972 revealed the presence of genotypes A and B in the country (Huang et al., Reference Huang, Cerenius and Söderhäll1994). In the UK, RAPD-PCR genotyping of A. astaci axenic culture isolated during a crayfish plague outbreak in 1990 in the River Arrow (Herefordshire, UK) (Lilley et al., Reference Lilley, Cerenius and Söderhäll1997; Alderman, Reference Alderman2003) and microsatellites analysis of crayfish samples from the Mochdre Brook (Powys) (James et al., Reference James, Nutbeam-Tuffs, Cable, Mrugala, Viñuela-Rodriguez, Petrusek and Oidtmann2017) revealed the presence of genotype group B only. Genotype group A has not been detected in the UK to date. Our more sensitive diagnostic primers allowed for the first detection of genotype group A in the UK co-existing with genotype group B in a single outbreak. While the presence of genotype groups and haplogroups A and B has often been reported in different sites and/or sampling occasions in the same geographical area, such as in Finland (Viljamaa-Dirks et al., Reference Viljamaa-Dirks, Heinikainen, Torssonen, Pursiainen, Mattila and Pelkonen2013), in Estonia (Kaldre et al., Reference Kaldre, Paaver, Hurt and Grandjean2017) and in Turkey (Kokko et al., Reference Kokko, Harlioglu, Aydin, Makkonen, Gökmen, Aksu and Jussila2018), the co-existence of different genotype groups and haplogroups in the same site/sample have been reported only rarely, examples includes Croatia (Maguire et al., Reference Maguire, Jelić, Klobučar, Delpy, Delaunay and Grandjean2016) and the Romanian Danube basin (Panteleit et al., Reference Panteleit, Keller, Diéguez-Uribeondo, Makkonen, Martín-Torrijos, Patrulea, Pîrvu, Preda, Schrimpf and Pârvulescu2018). As recent studies with microsatellite markers suggest the concomitant presence of different genotypes and genotype groups in the same geographical area (Grandjean et al., Reference Grandjean, Vrålstad, Diéguez-Uribeondo, Jelić, Mangombi, Delaunay, Filipová, Rezinciuc, Kozubíková-Balcarová, Guyonnet, Viljamaa-Dirks and Petrusek2014, Reference Grandjean, Roques, Delaunay, Petrusek, Becking and Collas2017; Kozubíková-Balcarová et al., Reference Kozubíková-Balcarová, Beran, Ďuriš, Fischer, Horká, Svobodová and Petrusek2014; Maguire et al., Reference Maguire, Jelić, Klobučar, Delpy, Delaunay and Grandjean2016; Kaldre et al., Reference Kaldre, Paaver, Hurt and Grandjean2017), the co-existence of multiple genotypes and genotype groups even within the same samples cannot be excluded. The lack of genotype group A detection in the UK until now could be explained by the difficulty of isolation and maintenance of A. astaci pure cultures in laboratory conditions (Oidtmann et al., Reference Oidtmann, Schmid, Rogers and Hoffmann1999) and that techniques allowing genotyping/haplotyping directly from infected animals have been developed only recently (Grandjean et al., Reference Grandjean, Vrålstad, Diéguez-Uribeondo, Jelić, Mangombi, Delaunay, Filipová, Rezinciuc, Kozubíková-Balcarová, Guyonnet, Viljamaa-Dirks and Petrusek2014; Makkonen et al., Reference Makkonen, Jussila, Panteleit, Keller, Schrimpf, Theissinger, Kortet, Martín-Torrijos, Sandoval-Sierra, Diéguez-Uribeondo and Kokko2018; Minardi et al., Reference Minardi, Studholme, van der Giezen, Pretto and Oidtmann2018), thus the detection of this genotype group in the UK could have been missed.
In conclusion, the present study demonstrates the possibility to develop informative molecular markers combining bioinformatics and molecular biology techniques to genotype the causative agent of crayfish plague, A. astaci. The assays developed are based on mtDNA and therefore more sensitive than any other genotyping markers are available in the literature. They can be applied to both pure cultures and crayfish tissue extractions from outbreak and carrier samples. Moreover, for the first time, we demonstrate the co-existence of A. astaci genotype groups A and B in UK waters.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182019000283.
Author ORCIDs
Diana Minardi, 0000-0002-9426-8627; David Studholme, 0000-0002-3010-6637; Mark van der Giezen, 0000-0002-1033-1335.
Acknowledgements
The authors thank Jiri Svoboda and Adam Petrusek for providing A. astaci isolate KB13. This work was conducted within the Centre for Sustainable Aquaculture Futures, a joint initiative between the University of Exeter and the Centre for Environment, Fisheries and Aquaculture Science (Cefas).
Financial support
This work was funded by a Cefas-Exeter University Alliance PhD Studentship to DM. The Exeter Sequencing Service was funded by a Wellcome Trust Institutional Strategic Support Award (WT097835MF).
Conflict of interest
The authors declare no conflicts of interest.
Ethical standards
Not applicable.