


INTRODUCTION
The biology of clinically important flagellated protozoan parasites from the genus Leishmania has been a subject of increasing academic interest. These parasites have a life cycle with two distinct forms: extracellular promastigotes that proliferate within the digestive tract of haematophagous female sandflies and intracellular amastigotes that thrive in the phagolysosomes of macrophages (Bates, Reference Bates2007; Mougneau et al. Reference Mougneau, Bihl and Glaichenhaus2011). Within susceptible mammalian hosts, the parasite proliferation and associated immune response originate a group of diseases known as leishmaniasis. Human leishmaniasis is characterized by different clinical presentations ranging from self-healing cutaneous manifestations to fatal visceralizing ailments (Kedzierski, Reference Kedzierski2010). Yearly, among an at-risk population of 350 million people, 2 million new cases are reported. As there is no vaccine, disease control relies on effective medical care. Although several therapeutic options are available, problems related to toxicity and resistance prevent adequate control and disease eradication (Kedzierski, Reference Kedzierski2010). In consequence, a better understanding of these protozoa is urgent to enable the development of safer and affordable therapeutic approaches.
A stable and reproducible source of microorganisms is essential to study Leishmania. In the wild a steady population of Leishmania is maintained by the sylvatic life cycle. In laboratory settings, promastigotes can be obtained from infected sandflies or from axenic cultivation. Biosecurity related problems and the lack of adequate facilities negate the generalized use of sandflies to study promastigotes (Volf and Volfova, Reference Volf and Volfova2011; Freitas et al. Reference Freitas, Parreiras, Duarte, Secundino and Pimenta2012). Notwithstanding, the axenic promastigotes are considered similar to those found in sandflies (Gossage et al. Reference Gossage, Rogers and Bates2003). On the other hand, axenic amastigotes are not considered biological surrogates for intracellular amastigotes (Rochette et al. Reference Rochette, Raymond, Corbeil, Ouellette and Papadopoulou2009; Pescher et al. Reference Pescher, Blisnick, Bastin and Spath2011). As a consequence, the bulk of the available information about Leishmania has been obtained from axenic promastigotes.
Novy–MacNeal–Nicolle (NNN) was the first medium used for the in vitro growth of Leishmania promastigotes (Nicolle, Reference Nicolle1908). Afterwards, several media were developed in a continuous effort to create more affordable and defined options for the growth of Leishmania (Hendricks et al. Reference Hendricks, Wood and Hajduk1978; Steiger and Black, Reference Steiger and Black1980; Palomino, Reference Palomino1982; Limoncu et al. Reference Limoncu, Balcioglu, Yereli, Ozbel and Ozbilgin1997; Merlen et al. Reference Merlen, Sereno, Brajon, Rostand and Lemesre1999; Singh et al. Reference Singh, Mohapatra and Sivakumar2000; Schuster and Sullivan, Reference Schuster and Sullivan2002; Muniaraj et al. Reference Muniaraj, Lal, Kumar, Sinha and Das2007; Sharief et al. Reference Sharief, Khalil, Omer and Abdalla2008; Tasew et al. Reference Tasew, Kebede, Wolday, Gadisa, Britton, Eidsmo and Akuffo2009; Rodrigues Ide et al., Reference Rodrigues Ide, da Silva, dos Santos, Vermelho, Alviano, Dutra and Rosa Mdo2010; Grekov et al. Reference Grekov, Svobodova, Nohynkova and Lipoldova2011). The most common options used for cultivation are semi-defined media supplemented with fetal calf serum (FCS) in concentrations varying between 5% and 20% (Schuster and Sullivan, Reference Schuster and Sullivan2002). FCS is expensive and highly variable in composition. Its replacement has long been considered an advance towards the standardization of in vitro cultivation. Some FCS substitutes such as milk (Muniaraj et al. Reference Muniaraj, Lal, Kumar, Sinha and Das2007) or urine (Armstrong and Patterson, Reference Armstrong and Patterson1994; Singh et al. Reference Singh, Mohapatra and Sivakumar2000) have been proposed but their use is limited. Years of axenic culture refinement have enabled the definition of generalized nutritional requirements permitting the creation of completely defined media (Steiger and Black, Reference Steiger and Black1980; Merlen et al. Reference Merlen, Sereno, Brajon, Rostand and Lemesre1999). The main nutritional requirements of Leishmania include: heme (Chang and Chang, Reference Chang and Chang1985); 6-hydroxymethylpterine and related pteridines (Bello et al. Reference Bello, Nare, Freedman, Hardy and Beverley1994); high levels of folic acid (Kar, Reference Kar1997); the vitamins thiamine, nicotinic acid, pantothenate, riboflavin and biotin (Schuster and Sullivan, Reference Schuster and Sullivan2002); and also several essential amino acids (Schuster and Sullivan, Reference Schuster and Sullivan2002). As consequence, the production of completely defined media is normally cumbersome, requiring more than 30 individual components (Steiger and Black, Reference Steiger and Black1980; Merlen et al. Reference Merlen, Sereno, Brajon, Rostand and Lemesre1999).
The traditional requisites for the development of new media for Leishmania spp. culture are sustained growth and infectivity of the parasites. Nonetheless, promastigote development in the insect vector involves several stages regulated by genetic and environmental factors (Bates, Reference Bates2007). The assumption that all developed media enable biologically identical promastigotes is therefore a dangerous oversimplification. In consequence, it is of crucial importance to evaluate if media can influence and modify promastigote biology. It is already known that the in vitro maintenance of Leishmania parasites rapidly leads to a decrease of virulence (Mitchell et al. Reference Mitchell, Handman and Spithill1984; Nolan and Herman, Reference Nolan and Herman1985; Moreira et al. Reference Moreira, Santarem, Loureiro, Tavares, Silva, Amorim, Ouaissi, Cordeiro-da-Silva and Silvestre2012). This phenotype has been related to a mounting incapacity to differentiate in the amastigote stage (Moreira et al. Reference Moreira, Santarem, Loureiro, Tavares, Silva, Amorim, Ouaissi, Cordeiro-da-Silva and Silvestre2012) or the loss of virulence factors through hypothetical medium adaptations (Mitchell et al. Reference Mitchell, Handman and Spithill1984; Pescher et al. Reference Pescher, Blisnick, Bastin and Spath2011). Overall, an ideal culture medium should not induce any bias capable of influencing experimental data and leading to experimental conclusions only valid for a specific combination of parasite and media. Human urine is a clear example of a media component with a specific biological imprint. When used as a medium supplement it increases parasite proliferation (Shamsuzzaman et al. Reference Shamsuzzaman, Furuya, Korenaga, Imamura and Hashiguchi1999) and infectivity (Allahverdiyev et al. Reference Allahverdiyev, Bagirova, Elcicek, Koc and Oztel2011).
Here, we evaluated the morphological, biological and immunological characteristics of Leishmania infantum promastigotes maintained in four conventional culture media, RPMI (McCarthy-Burke et al. Reference McCarthy-Burke, Bates and Dwyer1991), SDM 79 (Brun and Schonenberger, Reference Brun and Schonenberger1979), Schneider and NNN. Our approach enabled us to clearly assess significant alterations of relevant promastigotes’ biological parameters in each medium. Furthermore, we used the biological information obtained to develop a simple serum-free medium named cRPMI. cRPMI retains the biological characteristics required for becoming a medium of choice in the maintenance of these parasites.
MATERIALS AND METHODS
Parasites and cell culture
A cloned line of virulent L. infantum (MHOM/MA/67/ITMAP-263) was maintained at 27 °C in different culture media. SDM-79 (Brun and Schonenberger, Reference Brun and Schonenberger1979) (bSDM) was supplemented with 10% FCS (Lonza), 5 μg mL−1 hemin (Sigma), and 5 μ m biopterin (Sigma) – SDM. RPMI 1640 medium (Lonza) (bRPMI) was supplemented with 10% FCS, 2 mm L-glutamine (Lonza), 100 U mL−1 penicillin (Lonza), 100 mg mL−1 streptomycin (Lonza) and 20 mm HEPES buffer (Lonza) – RPMI. Schneider's Insect Medium (Sigma) was supplemented with 10% FCS, 200 units mL−1 penicillin, 200 units mL−1 streptomycin, 5 mm HEPES buffer, 2·5 g mL−1 Phenol Red (Sigma) – Schneider. NNN medium consists of 1·4% agar (Sigma), 0·6% NaCl (Merck), 31% defibrinated rabbit blood, 625 units mL−1 penicillin, 625 units mL−1 streptomycin and RPMI as liquid phase – NNN. Parasites were subcultured every 4 days with an initial inoculum of 106 parasites mL−1, except for SDM and Schneider in which the initial inoculum was 2·5×105 parasites mL−1. Promastigote to amastigote differentiation was achieved by culturing 107 stationary phase promastigotes mL−1 in a cell free culture medium at an acidic pH and 37 °C (MAA20) (Sereno and Lemesre, Reference Sereno and Lemesre1997). To minimize the possibility of clonal bias, we have performed three independent experiments, each one started with a distinct promastigote clone recovered from experimentally infected BALB/c mice.
Development of protein-free medium
FCS was passed through a centriprep Ultracel YM-3 filtering unit (Millipore) to remove the bulk of the protein content from the filtrate. The filtrate was then passed through the filtering device once again and then sterilized through a 0·2 μm filter (Millipore) and stored at 4 °C until use (this fraction will be herein referred as <3 kDa). The retentate was dialysed twice against phosphate buffered saline (PBS) to completely remove the low molecular weight components and stored at 4 °C until use (this fraction will be herein referred as >3 kDa). For the complementation experiments, L. infantum promastigotes growing in RPMI or SDM were washed twice in PBS and transferred for 2 days at a density of 106 mL−1 in the respective base medium (bRPMI or bSDM) complemented with one or combinations of the following supplements: 10% FCS; 10% >3 kDa; 10% <3 kDa or 2·5 μg mL−1 hemin. After 2 days, parasites were transferred again at a density of 106 mL−1 and evaluated for growth after 4 days by counting on a Neubauer chamber. To boost the growth obtained on bRPMI we added bSDM in defined percentages. For this, L. infantum promastigotes grown in RPMI were washed twice in PBS and used at a density of 106 mL−1 in bRPMI supplemented with 2·5 μg mL−1 of hemin and one or none of the following bSDM supplements: 10% bSDM, 20% bSDM, 50% bSDM or 100% bSDM. After 4 days of culture, the growth was determined using a Neubauer chamber. Morphology of the parasites was imaged using a Spinning Disk Confocal System Andor Revolution XD (ANDOR Technology, UK) using the 67× oil objective and treated using open source ImageJ software.
Growth curves
Promastigotes in bRPMI supplemented with 10% bSDM (cRPMI), RPMI, SDM, NNN or Schneider were grown for 24 h in a startup culture with the above defined initial inocula. These cultures were used as starter for the growth curves and followed for 7 days. At defined time points the parasites were counted using a Neubauer chamber.
Cell cycle analysis
Promastigotes were recovered from cultures in specific media at defined time points. Subsequently, parasites were washed twice and resuspended at a density of 2×106 in 1 mL of PBS 2% FCS. This was followed by the addition of 3 mL of cold absolute ethanol (Panreac) with continuous vortexing. Cells were then fixed for at least 1 h at 4 °C and washed twice in PBS. Before analysis, cells were resuspended in 50 μg mL−1 propidium iodide staining solution (Sigma) with 0·5 ng mL−1 RNase A (Sigma) and incubated 30 min at 4 °C. Data were collected in a BD FACSCalibur cytometer (20 000 gated events) and analysed by FlowJo software (Tree Star).
Viability analysis
Promastigotes were washed and resuspended at a density of 106 mL−1 in Annexin V binding buffer (BD Pharmingen). Parasites were then incubated at room temperature for 15 min with AnnexinV-Cy5 (BD Pharmingen) and 7-AAD (Sigma). Parasites subjected to ultraviolet light for 30 min and kept in culture for 4 h were used as a positive control. Data were collected in a BD FACSCalibur cytometer (20 000 gated events) and analysed by FlowJo software.
Real-time RT-PCR
Total RNA from 1×107 promastigotes was purified with TRIzol® reagent (Invitrogen), according to the manufacturer's instructions and resuspended in 10 μL RNase-free water. RNA concentration was determined by using a Nanodrop spectrophotometer (Wilmington) and quality was inspected for absence of degradation or genomic DNA contamination, using the Experion RNA StdSens Chips in the Experion™ automated microfluidic electrophoresis system (BioRad Hercules, CA, USA). RNA was stored at −80 °C until use. RT was performed with equal amounts of total extracted RNA (1 μg) obtained from parasites recovered from different experimental conditions by using Superscript II RT (Gibco BRL) and random primers (Stratagene). Real-time quantitative PCR (qPCR) reactions were run in duplicate for each sample on a BioRad My iCycler iQ5 in a 20 μL volume containing 4 μL of cDNA (diluted 25×), iQ SYBR Green Supermix (BioRad) and 500 nm (histone H4 and META1) or 250 nm (rRNA45 and SHERP) of primers. Specific primers for histone H4 (forward: 5′-ACACCGAGTATGCG-3′; reverse: 5′-TAGCCGTAGAGGATG-3′) (Moreira et al. Reference Moreira, Santarem, Loureiro, Tavares, Silva, Amorim, Ouaissi, Cordeiro-da-Silva and Silvestre2012), META1 (forward: 5′-GGGCAGCGACGACCTGAT-3′; reverse: 5′-CGTCAACTTGCCGCCGTC-3′) (Cunha et al. Reference Cunha, Carrillo, Sánchez, Cruz, Moreno and Cordeiro-da-Silva2013), Small Hydrophilic Endoplasmic Reticulum-associated Protein (SHERP) (forward: 5′-CAATGCGCACAACAAGATCCAG-3′; reverse: 5′-TACGAGCCGCCGCTTATCTTGTC-3′) (Moreira et al. Reference Moreira, Santarem, Loureiro, Tavares, Silva, Amorim, Ouaissi, Cordeiro-da-Silva and Silvestre2012) and rRNA45 (forward: 5′-CCTACCATGCCGTGTCCTTCTA-3′; reverse: 5′-AACGACCCCTGCAGCAATAC-3′) (Ouakad et al. Reference Ouakad, Bahi-Jaber, Chenik, Dellagi and Louzir2007) were obtained from Stabvida (Portugal), thoroughly tested and used for amplification. After amplification, a threshold was set for each gene and cycle threshold values (Ct values) were calculated for all samples. Gene expression changes were analysed using the built-in iQ5 Optical system software v2.1 (Bio-Rad laboratories, Inc). The results were normalized using as reference rRNA45 sequence (Ouakad et al. Reference Ouakad, Bahi-Jaber, Chenik, Dellagi and Louzir2007).
Staining of parasites with CFSE
Stationary-phase promastigotes (with 4 days of culture) diluted to a concentration of 1·2×107 mL−1 were used for carboxyfluorescein succinimidyl ester (CFSE, Invitrogen) labelling. Promastigotes were washed twice with PBS and labelled with 5 μ m of CFSE for 10 min at 37 °C, shaken every 3 min. Then, 9 mL of complete Dulbecco's modified Eagle's medium (DMEM, Lonza) was added and tubes were centrifuged for 10 min at 1200 g . The supernatant was discarded and the parasites were resuspended in 5 mL of complete DMEM and incubated at 4 °C for 5 min. After a final centrifugation for 10 min at 1200 g , the promastigotes were resuspended in complete DMEM before proceeding to macrophage infections.
In vitro macrophage infection
Cell suspensions of bone marrow were obtained by flushing the femurs and tibiae of 10–12-week-old BALB/c mice. The resulting cell suspension was cultured in DMEM supplemented with 10% heat-inactivated FCS, 2 mm L-glutamine, 100 U mL−1 penicillin, 100 U mL−1 streptomycin and 1 mm sodium pyruvate (Lonza). After overnight incubation at 37 °C, non-adherent cells were recovered (300 g for 10 min at room temperature) and cultured in 24-well culture dishes at a density of 106 cells mL−1. For macrophage differentiation, 10% L-929 cell conditioned medium was added to complete DMEM at days 0 and 4. At day 7 of culture, CFSE labelled promastigotes were incubated with the adherent bone marrow-derived macrophages at a 10 : 1 ratio. After 4 h, infection was stopped by washing the cells twice with PBS and further incubated for 24 and 48 h or immediately scrapped. The infection rates were determined at 4, 24 and 48 h post-infection by a BD FACSCalibur cytometer and analysed by FlowJo software as the percentage of CFSE+ cells.
Animal experiments and parasite quantification
Eight to twelve-week-old female BALB/c mice obtained from Instituto de Biologia Molecular e Celular (IBMC, Porto, Portugal) animal facilities were maintained in sterile cabinets and allowed sterile food and water ad libitum. Animal care and procedures were in accordance with institutional guidelines. All experiments were approved by and conducted in accordance with the IBMC.INEB Animal Ethics Committee and the Portuguese National Authority for Animal Health (DGAV – Direcção-Geral de Alimentação e Veterinária) guidelines. RS, JC and ACS have accreditations for animal research given by DGAV (Ministerial Directive 1005/92). Promastigotes recovered from stationary culture with 4 in vitro passages were collected, washed and suspended in sterile PBS. A volume of 200 μL of PBS containing 108 parasites was injected intraperitoneally. Mice of each group were euthanased at 56 days post-infection. The parasite burden in the spleen and liver was determined by limiting dilution as previously described (Silvestre et al. Reference Silvestre, Cordeiro-Da-Silva, Santarem, Vergnes, Sereno and Ouaissi2007).
Statistical analysis and division time calculations
The data were analysed using one-way ANOVA followed by Bonferroni's post-test for multiple comparisons when necessary. Minimum doubling time was calculated from experimental data using the algorithm provided by http://www.doubling-time.com/compute.php.
RESULTS
In vitro characterization of promastigotes grown in distinct media
We started by evaluating the biological characteristics of L. infantum promastigotes grown in different culture media. We performed daily evaluations of parasite number, viability and cell cycle analysis (Fig. 1A–C). Promastigote culture in SDM, Schneider and NNN enabled a rapid parasite growth that peaked at day 4 (from 1·0×108–1·3×108 parasites mL−1) followed by a steep decrease (Fig. 1A). This decrease in parasite numbers was concomitant with a significant diminution in viability and multiplicative parasites (Fig. 1A–C). In fact, the steep decrease in viability for SDM and Schneider required an adjustment of the starting inoculum to enable synchronous passages for all media (Fig. S1 – in Online version only). In contrast, the promastigotes cultured in RPMI presented a different growth profile with lower parasite densities (3×107 parasites mL−1) that were sustained for more than 10 days (data not shown). The maintenance of parasite density in RPMI was accompanied by high viability throughout the growth curve (>90%) and low numbers of multiplicative parasites (Fig. 1B and C). Concerning the cell cycle analysis, promastigotes in RPMI, SDM and NNN followed a trend of over time percentage decrease of S/G2 parasites (Fig. 1C and Figs S2 and S3 – in Online version only). In all these media, 40–50% of the parasites presented an S/G2 phenotype up to 2 days of growth. This was followed by a time-dependent decrease until less than 20% of the parasite population presented this phenotype. The only exception was in the Schneider medium where more than 50% of the population displayed a S/G2 cell cycle profile for all the days tested. The conjugation of growth curve and cell cycle analysis enabled the determination of minimum division time (Fig. 1D). SDM and Schneider media enabled smaller division times with an average of 6·56 h±0·89 and 5·89 h±0·90, respectively. The calculated division times for RPMI (9·74 h±0·65) and NNN (8·14 h±0·27) were significantly higher. To better integrate the biological information we also registered the dominant promastigote morphology at specific time points (Fig. 2). The first 2 days of culture were dominated by parasites with rounded bodies and flagella shorter than the body length. In fact, these represented the only detected form in the first day of culture. Cultures at day 4 displayed promastigotes with the traditional needle shape morphology (Fig. 2). The RPMI originated uniform promastigotes with long flagellum while the other media presented stationary cultures with mixed morphology. The NNN-grown parasites had a different growth phenotype with consistently smaller bodies than all the other media.
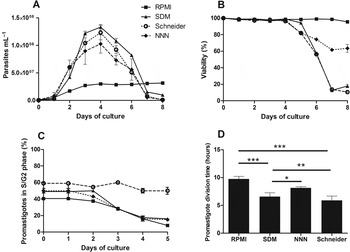
Fig. 1. Parameters of in vitro development of L. infantum in the different culture media. Promastigotes were cultured with an initial concentration of 1×106 mL−1 in RPMI and NNN or 2·5×105 parasites mL−1 in SDM and Schneider. (A) The growth curve was done by counting the parasites at different time points in a haemocytometer; (B) Promastigote viability was determined at the defined time points by quantifying the percentage of Annexin V (Ann V) and 7-aminoactinomycin D (7-AAD) negative promastigotes; (C) The percentage of S/G2 parasites was determined at different days; (D) Promastigote minimum division time calculated from the growth curves. *P<0·05; **P<0·01; ***P<0·001 as calculated by one-way ANOVA followed by Bonferroni's post-test analysed in Graph Pad Prism. The cell cycle and viability analysis was done using FACSCalibur cytometer and analysed by FlowJo software. The average of at least three experiments is shown.
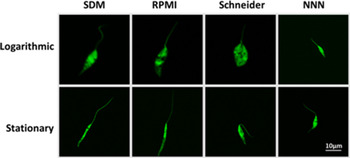
Fig. 2. Dominant morphology of logarithmic and stationary phase L. infantum. Promastigotes were cultured with previously established initial inoculum for each medium. After 1 or 4 days of culture CFSE-labelled parasites were mounted on CyGel™ and live confocal microscopy images were taken.
Evaluation of promastigotes’ infectivity
The evaluation of the in vitro infectivity was done with stationary phase parasites using bone marrow-derived macrophages as target cells (Fig. 3A and S4 – in Online version only). Similar percentages of infected cells were found after 4 h irrespectively of the medium used for promastigote maintenance (Fig. 3A). At 48 h post-infection, the SDM-grown parasites consistently presented a significantly higher level of infectivity (Fig. 3A). This increased in vitro infectivity did not translate in higher parasite burden in BALB/c mice parasite burden (Fig. 3B and C). On the contrary, the visceral parasite load induced by Schneider-cultivated parasites was significantly lower than the other tested media. The presence of metacyclic parasites and the capacity to differentiate into amastigotes have been described as important elements in virulence (Silvestre et al. Reference Silvestre, Cordeiro-Da-Silva, Santarem, Vergnes, Sereno and Ouaissi2007; Moreira et al. Reference Moreira, Santarem, Loureiro, Tavares, Silva, Amorim, Ouaissi, Cordeiro-da-Silva and Silvestre2012). Therefore, to better understand the differences in infectivity, we evaluated two parameters indicative of metacyclogenesis and the capacity to differentiate into axenic amastigotes. We analysed the expression of two metacyclogenesis-related genes, META1 and SHERP, and also Histone H4 (Fig. 4 and Fig. S5 – in Online version only) (Soto et al. Reference Soto, Quijada, Alonso and Requena1997; Ouakad et al. Reference Ouakad, Bahi-Jaber, Chenik, Dellagi and Louzir2007; Santos et al. Reference Santos, Silva, Zampieri, Lafraia and Floeter-Winter2011). A time-dependent fold increase in the relative expression of the above-mentioned genes relative to Histone H4 was detected. The greatest increase was detected for RPMI with a 40-fold increase for META1. In agreement with the in vivo data, Schneider-grown parasites presented no time-dependent variation in these genes. To confirm the possible enrichment of metacyclic parasites we seggregated the promastigote populations by Ficoll gradients (Yao et al. Reference Yao, Chen, Sudan, Donelson and Wilson2008). These gradients originated poor yields of metacyclic parasites, less than 6% of the original cultures, with no specific trend (data not shown). We also evaluated the differentiation into axenic amastigotes using MAA20 (Sereno and Lemesre, Reference Sereno and Lemesre1997). All media were capable of originating similar numbers of axenic amastigotes that were able to be subcultured (data not shown).

Fig. 3. In vitro and in vivo infectivity of promastigotes grown in different culture media. Parasites with four successive in vitro passages in SDM, RPMI, NNN or Schneider were used to perform in vitro and in vivo infections. (A) Bone marrow-derived macrophages were infected at a 1 : 10 (cell/parasite) ratio with CFSE-labelled promastigotes. Data were acquired in FACSCalibur cytometer and analysed by FlowJo software. Three independent experiments were performed and mean and standard deviation are shown; (B, C) BALB/c mice were infected with stationary phase promastigotes. After 6 weeks of infection, the parasite load was determined in spleen (B) and liver (C) by limiting dilution. The mean and standard deviation are shown. Two independent experiments were performed; one representative experiment is shown. *P<0·05; **P<0·01; ***P<0·001 as calculated by one-way ANOVA followed by Bonferroni's post-test analysed in Graph Pad Prism.

Fig. 4. Relative expression of metacyclogenesis-related genes in L. infantum promastigotes grown in the different media. Promastigote relative gene expression of META1 (A) and SHERP (B) determined by qPCR at the defined time points, when compared with Histone H4. Basic normalizations for the three genes were made using the reference gene rRNA45. Three independent experiments were done, each performed in duplicate; the mean of the three experiments is shown with SD.
Development of a FCS-free medium
After the initial analysis of general biological properties of the parasites grown in different media we developed a protein-free medium with a specific biological phenotype requisite: the conjugation of infectivity with long-lasting stationary cultures. Among the media evaluated, SDM and RPMI enabled the combination of these characteristics. The former allowed high yields of more infective but fast-dying parasites while the latter enabled long-lasting cultures. Although the media have different composition, the 10% FCS complementation was a common feature. Therefore, we evaluated the contribution of the FCS complementation. To achieve this, we used ultracentrifugation devices with 3 kDa membranes to separate the FCS into two distinct fractions: the retentate, composed of protein-associated components with a molecular weight higher than 3 kDa (>3 kDa) and the filtrate, composed of components with less than 3 kDa (<3 kDa). These two distinct fractions were then used to complement the defined SDM and RPMI bases (bSDM and bRPMI). Parasite growth in these complemented media was evaluated after 4 days of culture (Fig. 5A and B). The parasites were adapted for 2 days in the tested media to assess the real capacity for sustaining parasite growth. In all growth-capable conditions the parasite viability was higher than 90% allowing at least two subsequent passages (data not shown). Neither the bSDM nor bRPMI were able to promote growth even when complemented with <3 kDa. In contrast, the complementation with >3 kDa enabled total growth recovery for both media. In the latter, the parasites were indistinguishable from those in standard media (Fig. 1 and Fig. S6 – in Online version only). The bSDM and bRPMI, alone or upon complementation with <3 kDa presented limited growth, which was partially recovered by the addition of hemin, an essential component for Leishmania development (Chang and Chang, Reference Chang and Chang1985). The concentration of hemin was adjusted to 2·5 μg mL−1 as lower amounts were growth limiting (data not shown). Hence, hemin complementation in bSDM enabled 60% of normal growth (Fig. 5A), while in bRPMI it did not contribute towards significant growth (Fig. 5B). The complementation of both bases with <3 kDa and hemin induced distinct relative growths, 58% for bSDM and 27% for bRPMI. Significantly, the complementation of bRPMI with hemin and <3 kDa or hemin alone induced morphological features similar to RPMI-grown parasites (Fig. S6B – in Online version only). The same complementation for bSDM originated higher parasite yields with morphology that was different from SDM. To take advantage of the superior yield of bSDM and morphological characteristics provided by bRPMI, we combined bRPMI and bSDM (Fig. 5C). The combination that enabled the highest parasite yield with indistinguishable morphology from RPMI was obtained complementing bRPMI with 10% bSDM. This specific ratio of bSDM and bRPMI was called cRPMI. Also, the growth of other strains and species like Leishmania tarentolae was achieved upon adjustment of the bRPMI/bSDM ratio (data not shown). Another important characteristic for media development is the final cost. Thus, we compared the production cost of cRPMI with other standard media. cRPMI was the least expensive with half the cost of standard SDM (Table S1 – in Online version only) being ideal for parasite maintenance.

Fig. 5. Development of a protein-free medium for the growth of L. infantum promastigotes. Relative growth of L. infantum promastigotes (compared with SDM or RPMI) after 4 days of culture in bSDM (A) or bRPMI (B) complemented with: no supplement (NS); 10% of the lower fraction of serum (<3 kDa); 10% of high molecular components of serum (>3 kDa); 2·5 μg mL−1 hemin (Hem) and 10% of lower molecular components of serum with 2·5 μg mL−1 hemin (Hem; <3 kDa); 10% of high molecular components of serum with 2·5 μg mL−1 hemin (Hem; >3 kDa). One representative experiment of five (A) or three (B) is shown. (C) Growth of L. infantum promastigotes after 4 days of culture in bRPMI complemented with 2·5 μg mL−1 of hemin and different percentages of bSDM. The average of at least three independent experiments is represented.
In vitro characterization of promastigotes grown in cRPMI
The cRPMI enabled a continuous and reproducible growth with a maximum density of ≈1·2×107 parasites mL−1 (Fig. 6A). No growth defect was observed in long-term parasite maintenance (Fig. S7 – in Online version only). The cRPMI enabled up to 12 days of continuous culture with >90% viability, maintaining the subculture multiplicative capacity until day 9 of culture (Fig. 6A). Cell cycle analysis demonstrated that promastigotes grown in cRPMI also followed the general trend of decreasing the percentage of S/G2 parasites with less than 5% at day 5 (Fig. 6A). The division time for cRPMI was 12·09 h±0·41. Their morphology in cRPMI was indistinguishable from RPMI (Figs 2 and 6B). The fold increase of metacyclic genes was the highest among all tested media with more than 20-fold increase at day 5 for SHERP and 60-fold increase for META1. The cRPMI-grown parasites were also able to differentiate into axenic amastigotes in MAA20.

Fig. 6. Biology of L. infantum promastigotes in cRPMI. Promastigotes were cultured in cRPMI with an initial concentration of 1×106 mL−1. (A) The growth curve (solid line), percentage of S/G2 parasites (dark dashed line) and viability (light dashed line) were determined at different time points. Parasite number was determined using a haemocytometer. Promastigote viability was determined by the percentage of Annexin V (Ann V) and 7-aminoactinomycin D (7-AAD) negative promastigotes. The capacity of the parasites to originate effective inoculates (grey bars) was determined as follows: in each day of culture, an aliquot of parasites was recovered and used as inoculum to initiate new subpassages with 1×106 mL−1. The number of parasites present in culture 2 days after was counted with a haemocytometer. The cell cycle and viability analysis were done using FACSCalibur cytometer and analysed by FlowJo software. The average of three independent experiments is shown; (B) After 1 or 4 days of culture live confocal microscopy images were taken from the cRPMI-grown parasites stained with CFSE and mounted on CyGel™; (C) Promastigote relative gene expression of META1 and SHERP compared with histone H4 was determined by qRT-PCR at the defined time points. Normalizations for the three genes were made using the reference gene rRNA45. Three independent experiments were done, each performed in duplicate; the mean of the three experiments is shown with SD; (D) Bone marrow-derived macrophages were infected at a 1 : 10 (cell/parasite) ratio with CFSE-labelled promastigotes submitted to four successive in vitro passages in cRPMI. Data were acquired by FACSCalibur cytometer and analysed by FlowJo software. Three independent experiments were performed; one representative experiment is shown; (E) BALB/c mice were infected with stationary phase-promastigotes. Six weeks post-infection, the parasite load was determined in spleen and liver by limiting dilution. The mean and standard deviation are shown. Two independent experiments were performed; one representative experiment is shown. *P<0·05; **P<0·01; ***P<0·001 as calculated by one-way ANOVA followed by Bonferroni's post-test analysed in Graph Pad Prism.
Promastigotes grown in cRPMI maintained in vitro and in vivo infectivities without significant loss of virulence
The in vitro infectivity of promastigotes grown in cRPMI was evaluated as above (Fig. 6D). The infection profile was similar to the one obtained with RPMI-grown parasites with the number of infected cells dropping to less than 20% at 48 h. The in vivo infectivity was similar to RPMI, SDM and NNN (Figs 3 and 6E). We also evaluated the short-term loss of infectivity associated with continuous subculture (Moreira et al. Reference Moreira, Santarem, Loureiro, Tavares, Silva, Amorim, Ouaissi, Cordeiro-da-Silva and Silvestre2012). For this, we have made infections with promastigotes that were maintained in SDM, RPMI or cRPMI for 20 passages. The predicted number of generations for each passage (using 5 days of subculture) is higher in SDM (9 generations) when compared with RPMI (5 generations) and cRPMI (3·5 generations). Promastigotes subcultured in cRPMI medium maintained infectivity to similar levels as P4 promastigotes (Fig. 7). In opposition, all promastigotes grown in other tested media had a significant loss of infectivity after 20 in vitro successive passages. This loss was more evident in SDM-grown promastigotes that presented the most significant drop in infectivity (more than 50% – Fig. 7). A similar time-dependent loss of infectivity was already reported for RPMI (Moreira et al. Reference Moreira, Santarem, Loureiro, Tavares, Silva, Amorim, Ouaissi, Cordeiro-da-Silva and Silvestre2012).

Fig. 7. Overtime virulence loss. Bone marrow-derived macrophages were infected at a 1 : 10 (cell/parasite) ratio with CFSE-labelled promastigotes submitted to 4 or 21 successive in vitro passages in SDM, RPMI or cRPMI. Data were acquired by FACSCalibur cytometer and analysed by FlowJo software. Bars represent the percentage of infected cell by promastigotes with 21 passages in relation to the infection by parasites with 4 passages (dashed line); the mean and standard deviation of duplicates are shown. Three independent experiments were performed; one representative experiment is shown.
DISCUSSION
Ideally the use of distinct media should not be a determining factor in Leishmania biology, enabling comparison of experimental results obtained with promastigotes grown in different media. To evaluate the potential bias induced by distinct media, we analysed several promastigote characteristics in four distinct commonly used media. In a simplistic perspective, the development of promastigote forms can be divided into logarithmic (procyclic) and stationary stages. The correct definition of these stages is pivotal because the characteristics of multiplicative and stationary phase parasites are distinct. In fact, several studies are supported by the correct definition of these stages (Holzer et al. Reference Holzer, McMaster and Forney2006; McNicoll et al. Reference McNicoll, Drummelsmith, Muller, Madore, Boilard, Ouellette and Papadopoulou2006). Therefore, we started to define these two basic promastigote stages in the context of different media. All the promastigote growth curves were characterized by a phase of active increase in parasite number followed by a slower growth or stationary phase (Fig. 1A). The promastigote division time calculated for each individual media during the first 2 days was remarkably constant (data not shown). Still, inter-medium variations were detected ranging from 5·89 h±0·90 (Schneider) to 9·74 h±0·65 (RPMI) (Fig. 1D). This media dependency of the average division time was previously reported (Fritsche et al. Reference Fritsche, Sitz, Weiland, Breitling and Pohl2007) and will have an immediate impact in any study that evaluates cell cycle progression. For example, full cell cycle progression in Leishmania mexicana using M199-based medium was shown to take 7·1 h (Wheeler et al. Reference Wheeler, Gluenz and Gull2011). This value is similar to the division times for our strain in SDM (6·56 h±0·89), Schneider (5·89 h±0·90) but significantly different from the values obtained for RPMI (9·74 h±0·65) and NNN (8·14 h±0·27). The use of minimum division time to define the logarithmic phase suggests that the parasites are dividing with unrestrained speed for the first 2 days of culture. This is supported by cell cycle analysis, as for the first 2 days more than 40% of parasites were in S/G2 phase (Fig. 1C). Nonetheless, the use of the growth curve to define the time frame for recovery of stationary phase parasites can be misleading. Considering exclusively the promastigotes’ growth curve profile, for SDM, NNN and Schneider the logarithmic phase will be limited to the first 2 days and the stationary between day 3 and 5. Notwithstanding, the cell cycle profile of Schneider-grown parasites does not support this assumption. Promastigotes in Schneider presented an atypical behaviour with more than 50% being in a stable S/G2 phase throughout the culture span. This S/G2 profile suggests continuous division with the absence of a defined stationary phase as it is traditionally characterized. The over time increase in the theoretical division time indicates that cells take a longer time to complete the cell cycle – considering that cell death is negligible until day 4 (more than 90% viability) (Fig. 1B). The possibility of a reduced/absent stationary phase in Schneider parasites was reinforced by: morphological information that supported the existence of multiplicative parasites even at later stages of culture, no increase in metacyclic specific genes and lower in vivo infectivity in BALB/c mice (logarithmic parasites are known to be less infective; Alvarez-Rueda et al. Reference Alvarez-Rueda, Biron and Le Pape2009). The RPMI-grown parasites present a distinct growth behaviour from the other media, evaluated with a long stationary phase and high viability lasting until day 10. The cell cycle profile fully supports this assumption (Fig. 1C). Interestingly, the general characteristics of the parasites are maintained over time with no significant change in infectivity (data not shown). In consequence, simple generalizations of the growth phase of Leishmania cultures must be avoided. The proper definition of the time frame dominated by logarithmic or stationary phase parasites must be supported by cell cycle and growth curve derived information (including minimum division time and viability).
The capacity to infect and originate productive infections is a characteristic of stationary phase parasites. The SDM-grown parasites presented significantly higher infection levels after 48 h than the other tested media (Fig. 3). A higher number of immunomodulatory metacyclic parasites could explain this difference (Moreira et al. Reference Moreira, Santarem, Loureiro, Tavares, Silva, Amorim, Ouaissi, Cordeiro-da-Silva and Silvestre2012). However, neither the metacyclic specific gene expression nor the recovery from Ficoll gradient supported this possibility. Indeed, the SDM-grown parasites present similar metacyclic gene expression as RPMI parasites (Fig. 4 and Fig. S5 – in Online version only). Other factors can be accountable for these differences in infection. Apoptotic parasites have been shown to be relevant for the infectious process (Wanderley et al. Reference Wanderley, Pinto da Silva, Deolindo, Soong, Borges, Prates, de Souza, Barral, Balanco, do Nascimento, Saraiva and Barcinski2009). The level of apoptotic parasites was distinct among the cultures evaluated (Fig. 1B), with a steep increase in apoptotic cells after day 4 (except for RPMI). This might contribute to the differences found in SDM as it was shown for Leishmania braziliensis that apoptotic parasites inhibit the inflammatory response (Wanderley et al. Reference Wanderley, Pinto da Silva, Deolindo, Soong, Borges, Prates, de Souza, Barral, Balanco, do Nascimento, Saraiva and Barcinski2009). In fact, at day 4 the percentage of apoptotic parasites in SDM culture is higher, although not statistically significant, than in RPMI. Therefore, a significant experimental bias may be induced if day 5 parasites were used since parasite viability is significantly decreased in NNN, Schneider and SDM. Also, the promastigotes from Schneider presented a specific phenotype related to infection as they are significantly less infective in vivo. The lower expression of metacyclic specific genes hints at the possibility of having fewer metacyclic parasites. Still, we cannot infer that the differences in in vivo infectivity are directly related with a possible reduced metacyclogenesis. The attempts to recover metacyclic parasites from each media did not give additional significant information. Moreover, promastigotes cultivated in SDM, which are more resistant to macrophage-specific killing (Fig. 3A), do not express higher levels of those genes. These facts reinforce the importance of solid parasite characterization before the intended use. Interestingly, the reported differences for in vitro infections did not translate into significantly different in vivo infections for SDM, RPMI and NNN. The high number of parasites used and the infection route might account for the lack of differences between infections.
A possible explanation for the significant biological differences reported here might be the existence of different promastigote forms (Rogers et al. Reference Rogers, Chance and Bates2002). In the sandfly the promastigotes follow a specific developmental process encompassing the differentiation from amastigotes to metacyclic promastigotes. This process involves successive promastigote stages: procyclic, nectomonads, leptomonads and metacyclic (Rogers et al. Reference Rogers, Chance and Bates2002). These forms have distinct predicted biological roles and morphology (Bates, Reference Bates2008). The latter allows their distinction by simple microscopic analysis (Rogers et al. Reference Rogers, Chance and Bates2002). Interestingly, it was proposed that starting from amastigotes, one can find all these proposed developmental stages in vitro (Gossage et al. Reference Gossage, Rogers and Bates2003). The mechanisms responsible for these changes are not clear, although some facts related to the metacyclic development are known. Culture at low pH was associated with increased metacyclogenesis in L. mexicana (Bates and Tetley, Reference Bates and Tetley1993). Glucose consumption in Trypanosomatids induces a decrease of environmental pH due to the production of oxidized intermediates and organic acids (Van Hellemond et al. Reference Van Hellemond, Opperdoes and Tielens1998). Therefore, it is clear that the media content can influence not only the density of parasites but also the population composition. Interestingly, the dominant parasite morphology in the different media could be fitted in distinct morphological groups (Rogers et al. Reference Rogers, Chance and Bates2002). On the first day all cultures presented procyclic parasites with cell bodies bigger than flagella and of variable width (Fig. 2). After 4 days of culture the panorama was distinct. RPMI presented very homogeneous cultures of needle shape parasites longer than 12 μm and possessing long flagella that resembled nectomonads. Interestingly, the nectomonads are not multiplicative forms. This fact might explain the abnormal time span of the RPMI cultures (more than 10 days) compared with all other tested media (5 days). It is possible that RPMI promastigotes are mostly arrested in the nectomonads stage with very few advancing to the leptomonads stage. This would explain the low numbers of metacyclic parasites found for this strain in RPMI on previous works (Moreira et al. Reference Moreira, Santarem, Loureiro, Tavares, Silva, Amorim, Ouaissi, Cordeiro-da-Silva and Silvestre2012). All the other media promote cultures with mixed morphology at 4 days of culture. The NNN-grown parasites were visibly smaller than all the other parasites (Fig. 2). This was an intriguing fact that reinforces the importance of media selection. Noteworthy was also the continuous presence of multiplicative promastigotes in Schneider medium. The parasites with 4 days are also multiplicative but distinct from day 1, with longer flagella and slower division times. These might represent the second promastigote multiplicative form, the leptomonads. It is possible that Schneider-grown parasites are not able to pass this last developmental stage, failing to originate metacyclic parasites. Cultures with 4 days in SDM presented a mixture of parasites with morphologies similar to nectomonads, leptomonads and metacyclic parasites. It is still unclear whether these distinct morphological forms are associated with relevant biological phenomena in vitro, and further studies must be done to evaluate this. Still, the distinct biological properties of the cultures point to the pressing necessity for standardization of culture conditions. In fact, the use of clonal populations, as a result of parasite genetic modifications, might exacerbate the potential of the media to induce a significant biological imprint on the parasites (Mendez et al. Reference Mendez, Fernandez-Perez, Santin, De La Fuente, Cuquerella, Gomez-Munoz and Alunda2001). Taking into consideration the different growth profiles induced by the media and the associated costs (Table S1 – in Online version only) one can determine specific media applications. For example, SDM is the most cost-efficient medium, therefore should be the best option to produce parasite mass for DNA extraction. Furthermore, if swift and continuous parasite growth is required, such as limiting dilution assays in parasite load quantification, Schneider is a solid choice as it conjugates fast growth, enabling rapid and easy to read results (the medium changes colour due to acidification induced by parasite multiplication). However for applications requiring protein recovery, or other biological determinations, there is a strong possibility of media-induced phenotypes, as parasite growth and infectivity can be media specific.
The evaluation of the biological characteristics of the distinct media enabled the creation of a simple FCS-free media for the routine culture of L. infantum. The convenience of removing FCS from the media is clearly shown using RPMI. Other groups have reported different parasite densities and smaller division times (Limoncu et al. Reference Limoncu, Balcioglu, Yereli, Ozbel and Ozbilgin1997; Merlen et al. Reference Merlen, Sereno, Brajon, Rostand and Lemesre1999; Fritsche et al. Reference Fritsche, Sitz, Weiland, Breitling and Pohl2007). These growth variations described for the RPMI might be due to FCS intrinsic heterogeneity. Our personal observations support this hypothesis. We detected a density variation of 1×107 resulting from a change on the FCS lot (Fig. S8 – in Online version only). Therefore, we pursued the creation of a medium that enabled the growth of virulent parasites with uniform stationary morphology and long-lasting viability. These attributes are important for long-term protein-free studies, such as exoproteome recovery, where reduced cell death is an essential requisite. The RPMI media was the media that enabled this phenotype. In the absence of FCS the RPMI base (bRPMI) did not sustain any parasite growth. Therefore, it required a supplement to the bRPMI. In the other studied media only SDM conjugated a protein-free base (bSDM) with highly infective parasites. The SDM medium enabled short-lived cultures with mixed morphology reaching more than twice the culture density of RPMI. As both media were complemented with 10% FCS, the differences in culture profile must originate in the composition of the base media. Leishmania spp. are auxotrophic for heme, therefore they require an exogenous heme source (Chang and Chang, Reference Chang and Chang1985). The removal of the serum leads to the loss of haemoglobin as the main usable iron/heme source (Krishnamurthy et al. Reference Krishnamurthy, Vikram, Singh, Patel, Agarwal, Mukhopadhyay, Basu and Mukhopadhyay2005; Carvalho et al. Reference Carvalho, Cruz, Santarem, Castro, Costa and Tomas2009). To overcome this limitation we used hemin (Gholamhosseinian and Vassef, Reference Gholamhosseinian and Vassef1988; Srivastava et al. Reference Srivastava, Sharma, Kamboj, Rastogi and Pandey1997). The complementation experiments using fractionated FCS confirm that both bases were unable to promote parasite growth or survival in the absence of hemin (Fig. 5A and B). The fractionation of FCS suggests that the protein fraction is required for culture density. This is not unexpected because bovine serum albumin can be added as a media supplement improving parasite doubling times (Schuster and Sullivan, Reference Schuster and Sullivan2002). The incapacity of the <3 kDa fraction to promote growth restoration in bSDM suggested that bSDM contains all the nutrients (or equivalent) existing in the <3 kDa fraction. The morphology of the parasites was also used also as quality control. We used as criteria of normal morphology the elongated and pointed promastigotes with an anterior flagellum exceeding the body length. In fact, parasites with abnormal morphology are usually in stress conditions (Zangger et al. Reference Zangger, Mottram and Fasel2002). The parasites in bSDM presented abnormal morphology in the absence of protein supplementation (Fig. S6A – in Online version only). On the contrary, the parasites in bRPMI presented the classical morphology of stationary phase parasites regardless of the complementation (Fig. S6B – in Online version only). Remarkably, it seemed to be medium-specific elements that enabled acquisition and maintenance of the traditional stationary forms. As the essential nutrients in FCS could be substituted by bSDM, we used it to complement bRPMI boosting the culture density (Fig. 5C). Indeed, 10% bSDM complementation of bRPMI enabled the growth of parasites morphologically indistinguishable from parasites cultivated in RPMI (Figs 2 and 6B). The proportion of both bases was strain and species specific. We were able to cultivate the non-pathogenic L. tarentolae which presented optimal growth with 20% bSDM in cRPMI (data not shown). The cRPMI enabled lower parasite densities with longer division times of 12·9 h±0·41. The viability, cell cycle and infectivity retained the characteristics of RPMI. This was expected because the bRPMI fraction accounts for 90% of cRPMI. Significantly, the high viability sustained over time led to a longer life span similar to RPMI. This culture longevity is significant, enabling studies with long-term growth of promastigotes. Prolonged viability also minimizes the number of passages required for parasite maintenance. In fact, this is highly relevant for in vitro culture of Leishmania as virulence loss results from continuous cultivation (Moreira et al. Reference Moreira, Santarem, Loureiro, Tavares, Silva, Amorim, Ouaissi, Cordeiro-da-Silva and Silvestre2012). This loss of virulence reported is consistent with the number of generations passed since recovery of parasites from the mammalian host (Moreira et al. Reference Moreira, Santarem, Loureiro, Tavares, Silva, Amorim, Ouaissi, Cordeiro-da-Silva and Silvestre2012). In fact, in our subculture setting, 20 passages enable at least 80, 120 and 200 generations for parasites cultivated in cRPMI, RPMI and SDM, respectively. It would be expected that the effects of virulence loss might be diminished in cRPMI when compared with the other two strains. In fact, 20 passages induced a significant in vitro infectivity loss at 48 h after infection in SDM-grown parasites when compared with RPMI and cRPMI. The only media without significant virulence loss was cRPMI, confirming the effect of the passages in infectivity. Moreover, parasites in cRPMI could be successfully passed until day 9 of culture (Fig. 6A). Therefore, the number of generations should decrease dramatically if we subculture the parasites at day 9 (≈40 generations). This will result in an expected significant increase in the usable lifespan of the parasites between their recovery from mice and intended elimination due to virulence loss. The low cost associated with cRPMI also supports its use for culture maintenance, but the lower yield makes it the least cost-effective media. This is not a significant limitation of cRPMI, nonetheless it is not the best option for producing large numbers of parasites. Therefore, in this work, we clearly demonstrated that axenic cultivation of promastigotes induces specific biological, morphological and immunological biases in a media-specific manner. This fact will inherently influence experimental interpretation and warrants further investigation. We also developed a serum-free semi-defined medium, cRPMI, which sustains the growth of promastigotes biologically indistinguishable from parasites cultivated in RPMI. This medium enables the affordable maintenance of infective parasites for longer periods, increasing the subculturing time.
FINANCIAL SUPPORT
This work was funded by FEDER funds through the Operational Competitiveness Programme – COMPETE and by National Funds through FCT – Fundação para a Ciência e a Tecnologia under the projects FCOMP-01-0124-FEDER-011054 (PTDC/SAU-FCF/100749/2008), FCOMP-01-0124-FEDER-011058 (PTDC/SAU-FCF/101017/2008), FCOMP-01-0124-FEDER-019648 (PTDC/BIA-MIC/118644/2010) and CIHR operating grants to MO. RS was supported by Programa Ciência – financed by Programa Operacional Potencial Humano POPH – QREN–Tipologia 4.2 – Promoção do Emprego Científico, co-funded by Fundo Social Europeu and National funding from Ministry of Science, Technology and Higher Education (MCTES). NS, JC and DM were supported by fellowships from FCT code SFRH/BD/37352/2007, SFRH/BD/48626/2008 and SFRH/BD/91543/2012, respectively. MO holds the Canada Research Chair in Antimicrobial Resistance.
SUPPLEMENTARY MATERIAL
To view supplementary material for this article, please visit http://dx.doi.org/10.1017/S0031182013001388.









