Introduction
Cryptosporidium, an apicomplexan protozoan parasite that belongs to the class Conoidasida is considered as the primary causative organism of respiratory and gastrointestinal cryptosporidiosis. This disease involves watery diarrhoea with or without a persistent cough in both immunocompetent and immunodeficient individuals. The parasite is capable of completing its lifecycle within a single host, resulting in cyst stages that are excreted in feces or through coughing and are capable of transmission to a new host. In children, cryptosporidiosis has been associated with impairment in growth, physical fitness and cognitive function and has been identified as the leading global cause of diarrheal mortality among infants aged between 12 and 23 months (Desai et al. Reference Desai, Sarkar and Kang2012). A recent study has suggested that infection with Cryptosporidium parvum could lead to digestive carcinogenesis in humans (Benamrouz et al. Reference Benamrouz2014). Cryptosporidiosis has emerged as a major cause of diarrhoeal disease and death in infants (Snelling et al. Reference Snelling2007). It essentially spreads through the fecal–oral route, often through contaminated water and food (Efstratiou et al. Reference Efstratiou, Ongerth and Karanis2017). Infections by Cryptosporidium parasites are estimated to be around 750 000 in the USA and around 500 million annually in developing countries (Scallan et al. Reference Scallan2011; Samie et al. Reference Samie2015). Though cryptosporidiosis is usually a self-limiting illness in healthy individuals and lasts on average up to 9–15 days; while in immunocompromised individuals, it can be life-threatening as there is no fully effective drug treatment. Treatment strategies are extremely limited as no vaccine is available for this parasite and nitazoxanide (NTZ) is the only FDA approved drug for cryptosporidiosis that shows moderate efficacy in immuno-compromised individuals (Abubakar et al. Reference Abubakar2007). Moreover, actual quantification of zoonotic and anthroponotic transmission of the pathogen is very difficult in the natural environment. Lack of long-term maintenance in cell culture and in vitro genetic modification difficulties in Cryptosporidium genome are also hampering the fast development of new therapeutic strategies for the parasite. Hence, there is a pressing need for alternative search strategies like computational approach to exploit the genomics data for identification of novel therapeutic candidates. In humans, Cryptosporidium hominis and Cryptosporidium parvum are two epidemiologically important diarrhoeal pathogens, which cause cryptosporidiosis.
In this study, an attempt has been made by comparative genomics and immunoinformatics approach to identify potential drug and vaccine candidates against C. parvum proteome. The proposed target discovery pipeline is largely independent of experimental data and the essentiality of proteins of interest which are non-homologous to the human host can be predicted if the protein is found in C. parvum and other cryptosporidium parasite proteomes (Ludin et al. Reference Ludin2012). Other in silico screening strategies like subcellular localization, drug target prioritization parameters and MHC binding potential were used for screening of novel therapeutic candidates.
Materials and methods
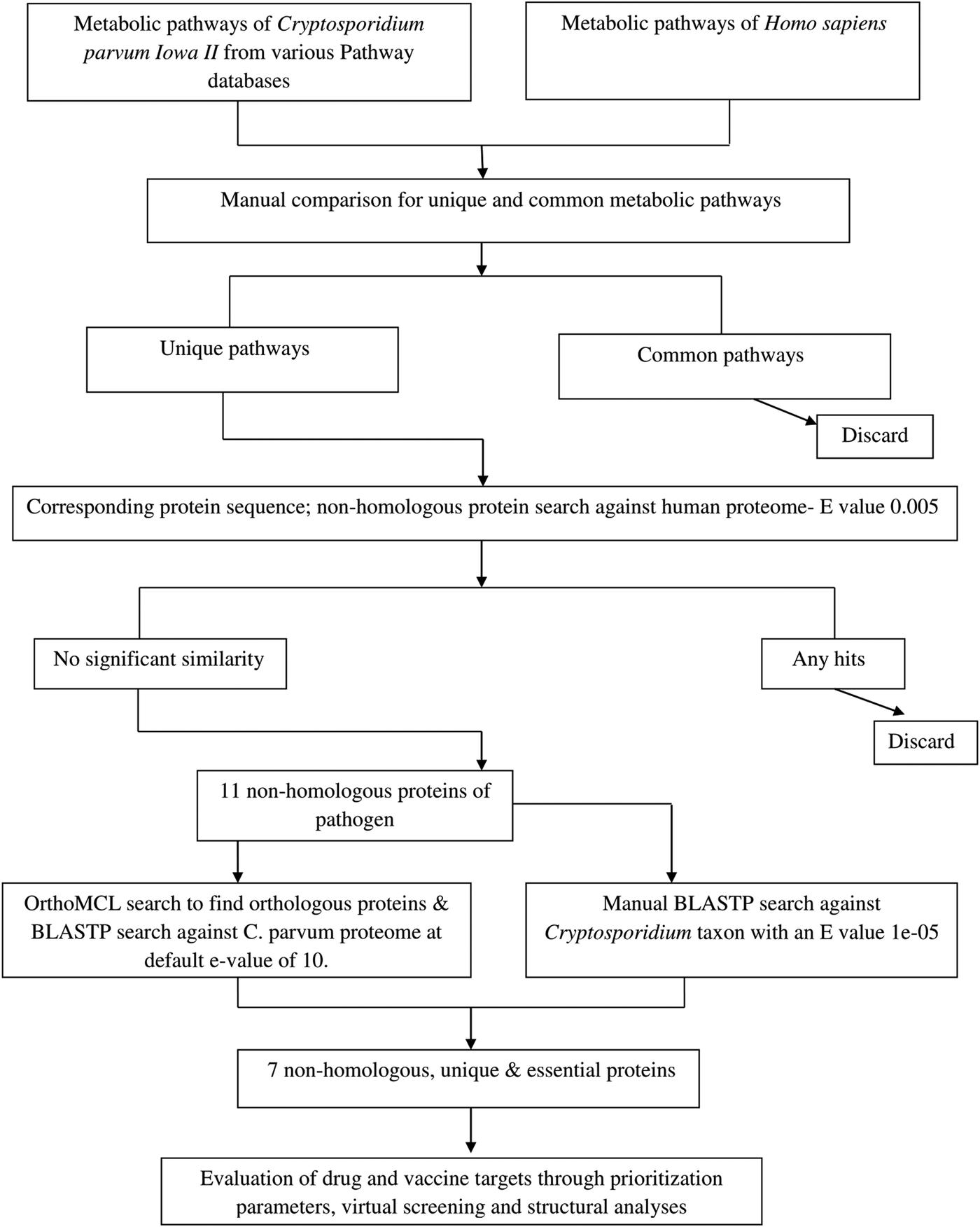
A systematic workflow was defined that involved several levels of filtration and identified the drug and vaccine targets in C. parvum genome using bioinformatics tools and methodology (Fig. 1).
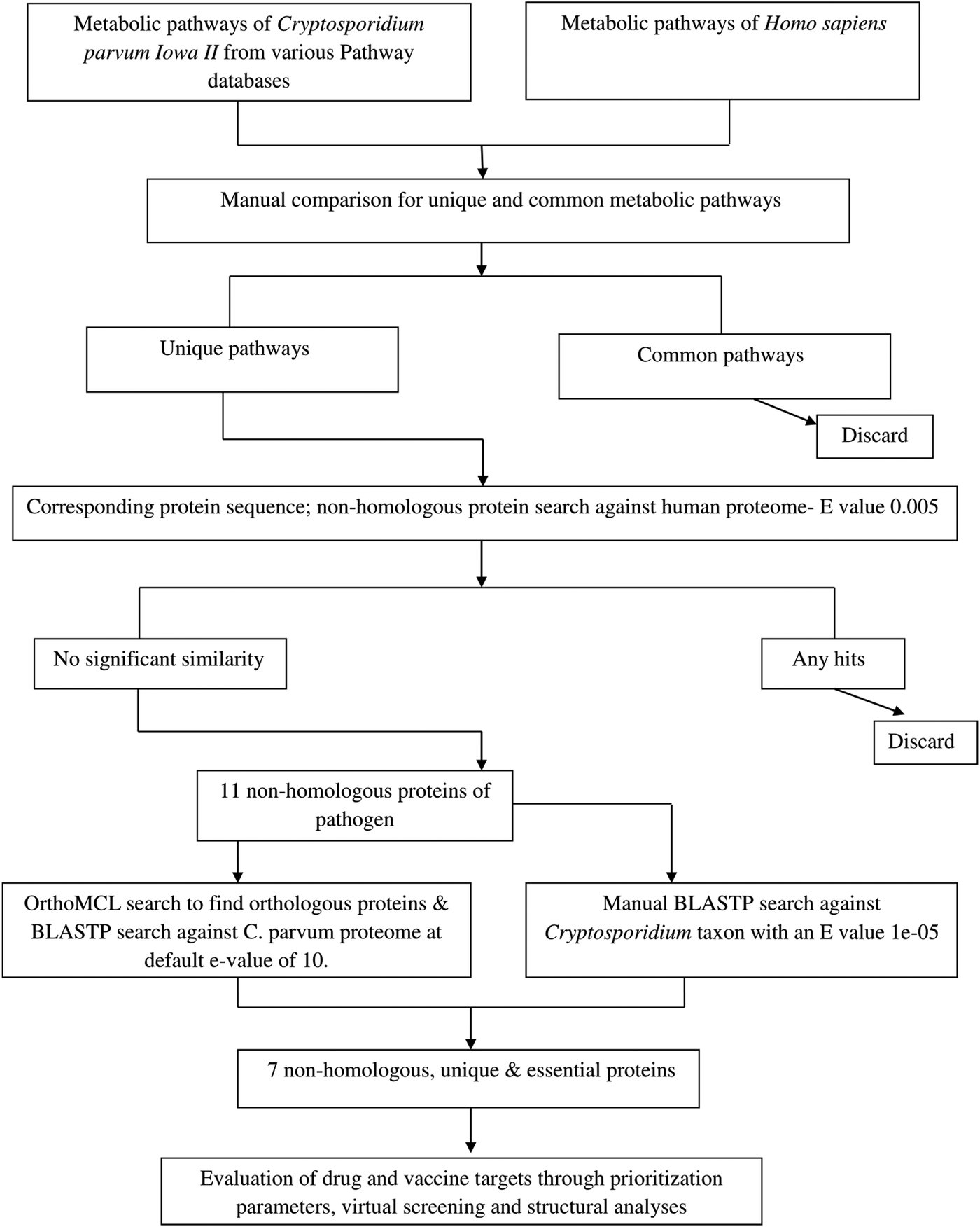
Fig. 1. Schematic representation of steps involved in drug target and vaccine candidate identification of Cryptosporidium parvum proteome through computational methods.
Identification of pathogen's unique metabolic pathways
All the currently curated metabolic pathways of C. parvum (Cpv) along with that of the human host were retrieved from the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database (Kanehisa et al. Reference Kanehisa2017) and a manual comparison was performed to find common and unique pathways in Cpv. In a similar manner, the comparison was performed between all currently available pathways of Cpv and human deposited at BioCyc pathway database (Caspi et al. Reference Caspi2016). Additionally, we have also used the Library of Apicomplexan Metabolic Pathways (LAMP) database version 2.0 (Shanmugasundram et al. Reference Shanmugasundram2012) for comparison purpose. Metabolic pathways present in both human and Cpv were considered as common pathways, whereas those present only in Cpv, but not in human were considered as unique pathways. The protein sequences of all the corresponding involved genes for each unique pathway generated from the above three approaches were also retrieved from the UniProt database (The UniProt Consortium, 2016). All the collected protein sequences were organized and were passed through further proteome subtractive channel.
Mining of non-homologous proteins to the human proteome
The collected protein sequences of unique pathways of the pathogen from the above step were subjected to NCBI BLASTP search with an e-value threshold set to 0.005 against human proteome. Proteins showing ‘no significant similarities’ or ‘no hits’ were considered as non-homologous proteins to the host proteome (Anishetty et al. Reference Anishetty, Pulimi and Pennathur2005; Ghosh et al. Reference Ghosh2014). Since non-homologous proteins minimize the chances of side effects and cross-reactivity caused by antibiotics, hence they can effectively serve as therapeutic candidates.
Essentiality assessment of Cpv proteins
Essential proteins of a cellular organism are the proteins which are necessary to survive and replicate. Hence, these proteins can be treated as desirable targets for antimicrobial treatments. Proteins can be essential if (i) they are orthologues in other Cryptosporidium species and (ii) have no other match of proteins in C. parvum proteome (Ludin et al. Reference Ludin2012). We have followed a three-step approach for essentiality prediction.
OrthoMCL (Li et al. Reference Li, Stoeckert and Roos2003) was employed to search for orthologous protein sequences in two other related Cryptosporidium species namely C. hominis strain: TU502 and C. muris strain: RN66, with a BLAST e-value of 1e−5. Furthermore, we performed a manual NCBI BLASTP search with an e-value of 1e−5 against Cryptosporidium taxon proteomes which includes that from C. hominis, C. muris, C. andersoni and C. ubiquitum. In the third and last step, those protein sequences were subjected to manual BLASTP search at a default e-value threshold of 10 against C. parvum proteome for no match. Finally, the proteins screened out from the above three-level steps were consider as non-homologous, conserved and essential proteins of C. parvum.
Drug target prioritization and druggability analysis
The proteins that are non-homologous to the human host and are essential for Cpv can be treated as potential drug targets. However, drug target selection can be constricted through the prioritization of screened proteins by following various structural and molecular criteria (Agüero et al. Reference Agüero2008). In our study, we have focused on parameters such as molecular weight calculation, subcellular localization of the targets, transmembrane domain prediction via TMHMM server v. 2.0 (Krogh et al. Reference Krogh2001), druggability test by DrugBank and availability of solved three-dimensional (3D) structures in the PDB database (Bernstein et al. Reference Bernstein1977) and Mod base (Pieper et al. Reference Pieper2013).
Subcellular localization prediction was made using CELLO v. 2.5 (Yu et al. Reference Yu2006) for classifying the predicted proteins as potential drug targets or vaccine candidates. Resultant datasets of CELLO v. 2.5 were further cross-checked with ESLPred2 server (Garg and Raghava, Reference Garg and Raghava2008). Identifying a similar protein that binds to the drug-like compound is regarded as the most reliable way to identify the druggability of a protein (Hajduk et al. Reference Hajduk, Huth and Tse2005). Hence, all identified potential drug targets were searched against the DrugBank database (Knox et al. Reference Knox2011). Hits found with DrugBank were considered as druggable targets whereas the remaining were considered as novel drug targets which further need to be validated experimentally.
Binding to MHC class I and II proteins
During an attack on foreign antigens, MHC class I and II play a significant role in the activation of cytotoxic T cells and helper T cells, respectively. ProPred and ProPred1 servers were used to predict MHC class II and MHC class I binding regions in the protein sequence, respectively. ProPred and ProPred1 servers allow locating loose binding sites of nine residue-long peptide regions on a protein that can bind to 51 different MHC class II alleles and 47 MHC class I alleles, respectively (Singh and Raghava, Reference Singh and Raghava2001, Reference Singh and Raghava2003). The percentage of best scoring natural peptides or the threshold was kept at its default value of 3% for ProPred and 4% for ProPred1 to maintain stringency. The plasma-membrane proteins from the previous step, which were proposed as the vaccine candidates, were submitted to these servers separately. A cut-off score was kept and for all alleles, proteins with a binding score above that cut-off were listed for each MHC category.
Additional characterization parameters
Certain additional protocols such as SignalP v. 4.1, VaxiJen, IgPred and AlgPred were also employed for characterization of the proteins which in our case will serve as probable vaccine candidates. SignalP v. 4.1 predicts the presence and location of signal peptide cleavage sites in residues of the proteins with server-recommended cut-off values of 0.45 and 0.50, optimized for correlation (Petersen et al. Reference Petersen2011). IgPred is used for prediction of different types of B-cell epitopes that can induce different classes of antibodies such as IgG, IgE and IgA (Gupta et al. Reference Gupta2013). VaxiJen web server is used for classifying a protein sequence as probable antigens above the user-specified threshold value of 0.5 (Doytchinova and Flower, Reference Doytchinova and Flower2007). AlgPred predicts whether the protein is a potential allergen or not at SVM threshold value of −0.4 (Saha and Raghava, Reference Saha and Raghava2006a).
Prediction of antigenic epitopes
Presence of epitopes or antigenic sites on proteins is vital for the development of synthetic peptide vaccines, immunodiagnostic tests and antibody production. The proposed vaccine candidate protein in the previous step was considered for prediction of B-cell epitopes and cytotoxic T lymphocyte epitopes by ABCpred (Saha and Raghava, Reference Saha and Raghava2006b) and CTLPred (Bhasin and Raghava, Reference Bhasin and Raghava2004), respectively. For our case, the length of the linear B cell epitopes was fixed at 16 and the cut-off was kept at 0.51. For nine-residue-long T cell epitope prediction, in the CTLPred server, the cut-off score for ANN was kept at 0.51, and for SVM it was set to 0.36, above which peptides are shown to be antigenic. Furthermore, DISOPRED2 was also used to check if the epitopes lay in unstructured regions of the protein (Ward et al. Reference Ward2004).
Homology modelling – 3D structure prediction and assessment
The homology models of the probable drug target protein and the top scoring epitopes on the proposed vaccine candidate protein for C. parvum were built using SWISS-MODEL (Biasini et al. Reference Biasini2014) and PEP-FOLD (Shen et al. Reference Shen2014), respectively.
The protein model was visualized in PyMOL software of Schrödinger Inc. (Schrödinger, Reference Schrödinger2010). The quality of the refined model was verified using SAVES meta-server (Structure Analysis and Verification Server) which comprises VERIFY3D (Eisenberg et al. Reference Eisenberg, Lüthy, Bowie, Carter and Sweet1997), PROCHECK (Laskowski et al. Reference Laskowski1993), ERRAT (Colovos and Yeates, Reference Colovos and Yeates1993) and PROVE modules (Pontius et al. Reference Pontius, Richelle and Wodak1996). PROCHECK examines the stereo-chemical quality of the generated protein structures by using the Ramachandran plot. The probable function of these proteins can be determined from the InterPro database (Finn et al. Reference Finn2016).
Binding site prediction, lead identification, virtual screening and docking studies
We have used the COFACTOR server (Roy and Zhang, Reference Roy and Zhang2012) for generating ligand-binding sites and for prediction of the lead ligand. The predicted binding site residues were further validated by 3DLigandSite server (Wass et al. Reference Wass, Kelley and Sternberg2010). Using the lead compound predicted by the COFACTOR server, different analogues of the same were searched from publicly available drug depository databases, namely, the ZINC database (Sterling and Irwin, Reference Sterling and Irwin2015), PubChem database (Kim et al. Reference Kim2016), ChEMBL database (Gaulton et al. Reference Gaulton2017) and anti-cryptosporidiosis compounds reported in Pathogen Box (Bessoff et al. Reference Bessoff2014). After removing duplicates and preliminary processing, compounds were all set for docking with the proposed drug target protein using the Lead Finder software (Stroganov et al. Reference Stroganov2008), implemented in Flare software package of Cresset-group (Cresset®, 2006), which uses the classical genetic algorithm with various local optimization procedures. The protein–protein docking study for the predicted epitopes on the probable vaccine candidate protein was performed by ClusPro web server (Kozakov et al. Reference Kozakov2017), which uses Fast Fourier transform-based PIPER for rigid body docking.
Results
Identification of metabolic pathways for Cpv
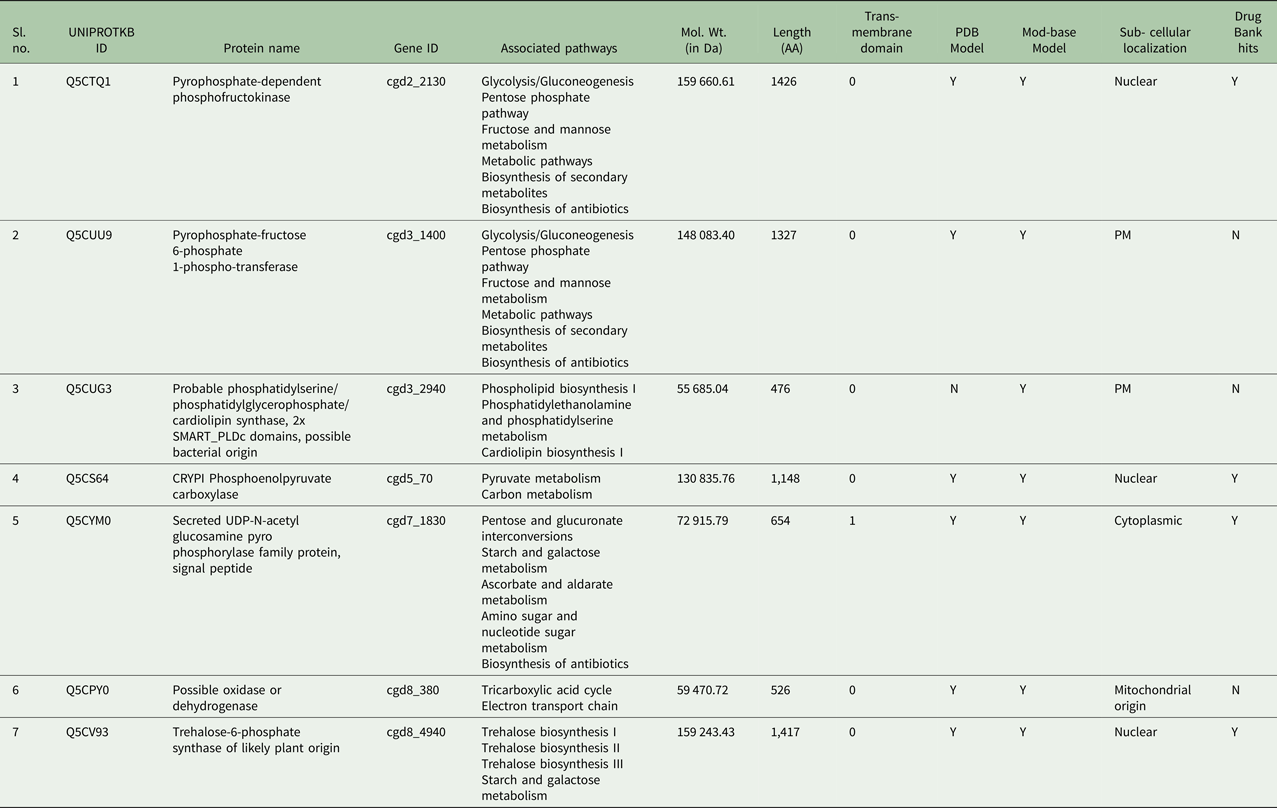
To begin the first step of finding potential therapeutic drug targets and vaccine candidates of C. parvum using the computational approach, initially, after retrieving 78 metabolic pathways of Cpv and 320 pathways of human host from the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database, a manual comparison was performed to find common and unique pathways in Cpv. This reported a total of 74 common pathways, four unique pathways in Cpv and 53 associated genes with the four unique pathways (Supplementary Table 1). Similarly, the comparison was performed independently between, 82 pathways of Cpv and 314 pathways of human deposited at BioCyc pathway database, which reported 31 common pathways, 51 unique pathways in Cpv and a total of 52 genes associated with the unique pathways after removing redundancies (Supplementary Table 2). Furthermore, 33 Cpv pathways deposited at Library of Apicomplexan Metabolic Pathways (LAMP) database version 2.0 were also compared with human proteome, which reported a total of 31 enzymes and 33 genes associated with the unique pathways of Cpv (Supplementary Table 3). After duplicates removal, a total of 103 protein sequences were reported from all the unique pathways found by comparison in the three databases. The 103 protein sequences were retrieved from the UniProt database.
Identification of non-homologous and essential proteins of Cpv
The proteins not having any significant similarity with human proteome are usually referred to as ‘non-homologous’ proteins. From the BLASTP search against human proteome at an e-value of 0.005, 11 proteins out of the 103 proteins were identified as non-homologous and were subjected to further screening procedures (Supplementary Table 4).
A total of 11 proteins were examined for their essentiality for parasite survival. As the DEG (Database of Essential Genes) did not provide much information about essential proteins in Cryptosporidium, we have followed a three-step approach previously described for essentiality assessment.
OrthoMCL search and NCBI BLASTP against Cryptosporidium taxon reported eight out of the 11 non-homologous proteins (with gene id: cgd2_2130, cgd3_1400, cgd3_2940, cgd5_70, cgd5_4440, cgd7_1830, cgd8_380 and cgd8_4940) were found in all five Cryptosporidium species (Supplementary Table 4). After a BLASTP search against C. parvum proteome with default e-value, seven proteins out of the eight proteins (cgd5_4440 protein was discarded as the e-value of search result was not found to be 0.0) were found to be having no other match in the parasite proteome. Hence the remaining seven proteins were considered for the next screening steps (Table 1).
Table 1. Non-homologous and essential proteins of C. parvum from UniProtKB with reference to humans as potential drug and vaccine targets from unique pathways
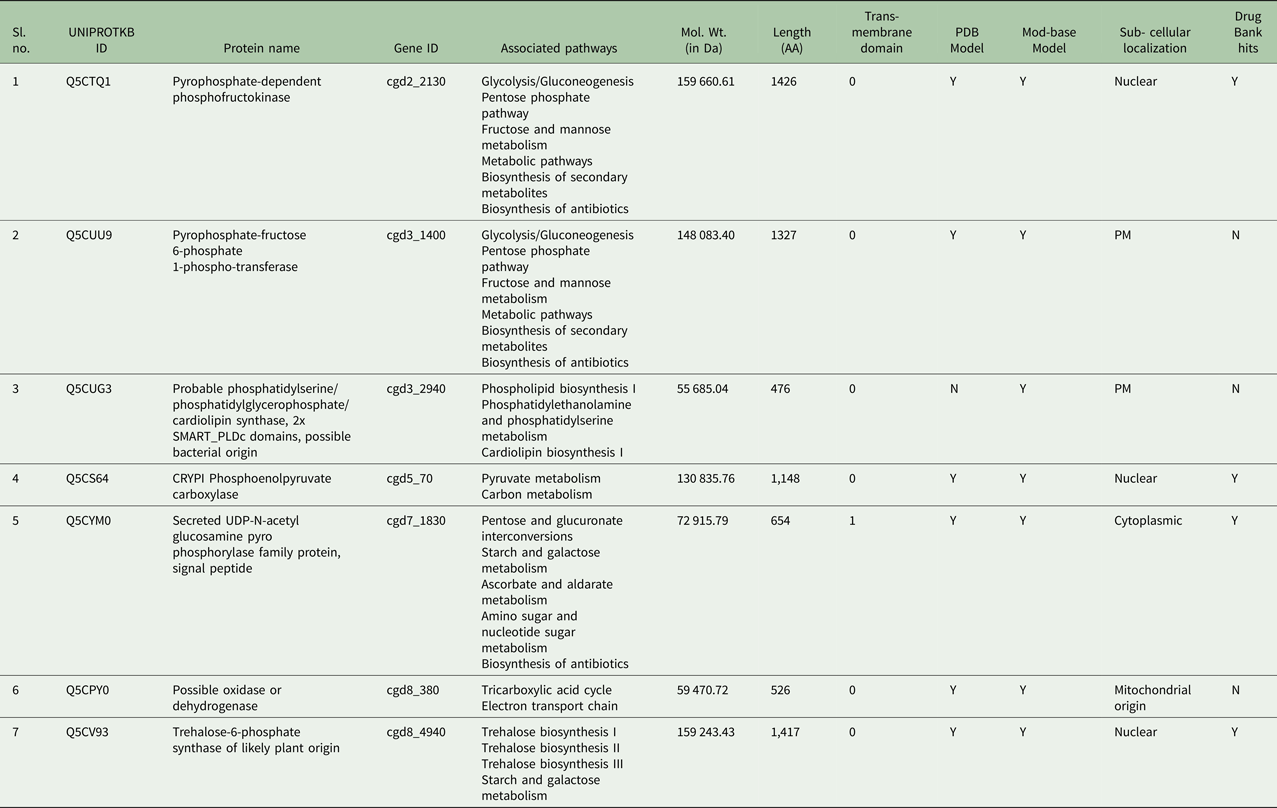
The sub-cellular localization is based on the consensus results through predictions by CELLO v 2.5 and ESLpred2. Trans-membrane helices data from TMHMM web-server.
Subcellular localization and drug target prioritization
The subcellular localizations of proteins, estimated through consensus results produced by ESLPred2 and CELLO eukaryotic subcellular localization prediction servers, determined three proteins (cgd2_2130, cgd5_70 and cgd8_4940) as nuclear, two as plasma membrane (cgd3_1400, cgd3_2940), one (cgd7_1830) as cytoplasmic and one (cgd8_380) as of mitochondrial origin. Several other characteristics of the selected seven proteins were listed in Table 1.
Cytoplasmic or membrane localization of the targets indicates they are likely to be easily purified for experimental studies. Proteins with low molecular weight (<100 kDa) increase the accessibility value of target protein (Butt et al. Reference Butt2011). Hence, we have considered the cytoplasmic protein cgd7_1830 as a drug target.
Affinity of protein antigens to MHC class I and II proteins
The proteins from the above calculations which were found to be located in the plasma membrane (cgd3_1400 and cgd3_2940), were subjected to ProPred1 and ProPred servers separately. The cut-off was kept at 90% on a log scale for MHC class I alleles and 70% for MHC class II alleles.
It was observed that, in ProPred1, protein cgd3_2940 (UniProtKB entry Q5CUG3) had two peptides in 100% range (with MHC alleles HLA-B*2705 and MHC- Kd) and two peptides in 90–100% range (with MHC alleles HLA-B*51 and HLA-B*5301), while in ProPred, it had 7 peptides in 70–80% range (with MHC alleles DRB1_1120, DRB1_1302, DRB1_1304, DRB1_1304, DRB1_1327, DRB1_1328, DRB1_1301). Similarly in ProPred1, protein cgd3_1400 (UniProtKB entry Q5CUU9) had two peptides in 100% range (with MHC alleles HLA- A20 Cattle and MHC- Kd) and two peptides in 90–100% range (with MHC alleles HLA-B*51 and HLA-B*5301), while in ProPred, it had only one peptides in 70–80% range (with MHC allele DRB1_1304) (Supplementary Table 5 and 7).
Additional characterization parameters
Several other protocols were also employed to characterize the proteins selected as vaccine candidates (cgd3_1400 and cgd3_2940). SignalP v.4.1 predicted that both the proteins have no signal peptide. IgPred conveyed that both can raise IgG antibodies. SVM-based AlgPred predicted that the protein cgd3_1400 is not a source of allergen with a score of −0.603 less than the threshold value of −0.4. However, the protein cgd3_2940 was predicted to be a probable source of allergen with a score of 0.348 more than the threshold value of −0.4. VaxiJen analysis of cgd3_1400 reported it to be non-antigenic with a score of 0.485, less than the parasite threshold value of 0.5, whereas that for cgd3_2940 reported a value of 0.529, more than the parasite threshold value; hence, it is a potential antigen (Supplementary Table 6). Hence, we have considered the membrane protein cgd3_1400 as a potential vaccine candidate.
Prediction of B cell and cytotoxic T cell epitopes
ABCpred predicted three linear B-cell epitopes on cgd3_1400 with a score of ⩾0.95. The highest ranking epitope was predicted to be HQMIETCIGFDSVTKS; having a score of 0.97, with starting position at 850th residue of the protein (Supplementary Table 8). CTLPred predicted three cytotoxic T-cell epitope on cgd3_1400, with YANPGPIQY as the highest ranking epitope; starting at 1202nd residue of the protein (Supplementary Table 8). Using DISOPRED2, it was found out that 21st, 26th, 566–602, 752–783, 919th, 920th, 925th, 926th, 983–997, 1140–1166, 1274–1327 residues of the protein sequence cgd3_1400 are disordered and none of the predicted top epitopes lay in disordered regions.
Homology modelling of protein and design of the 3D peptide structure
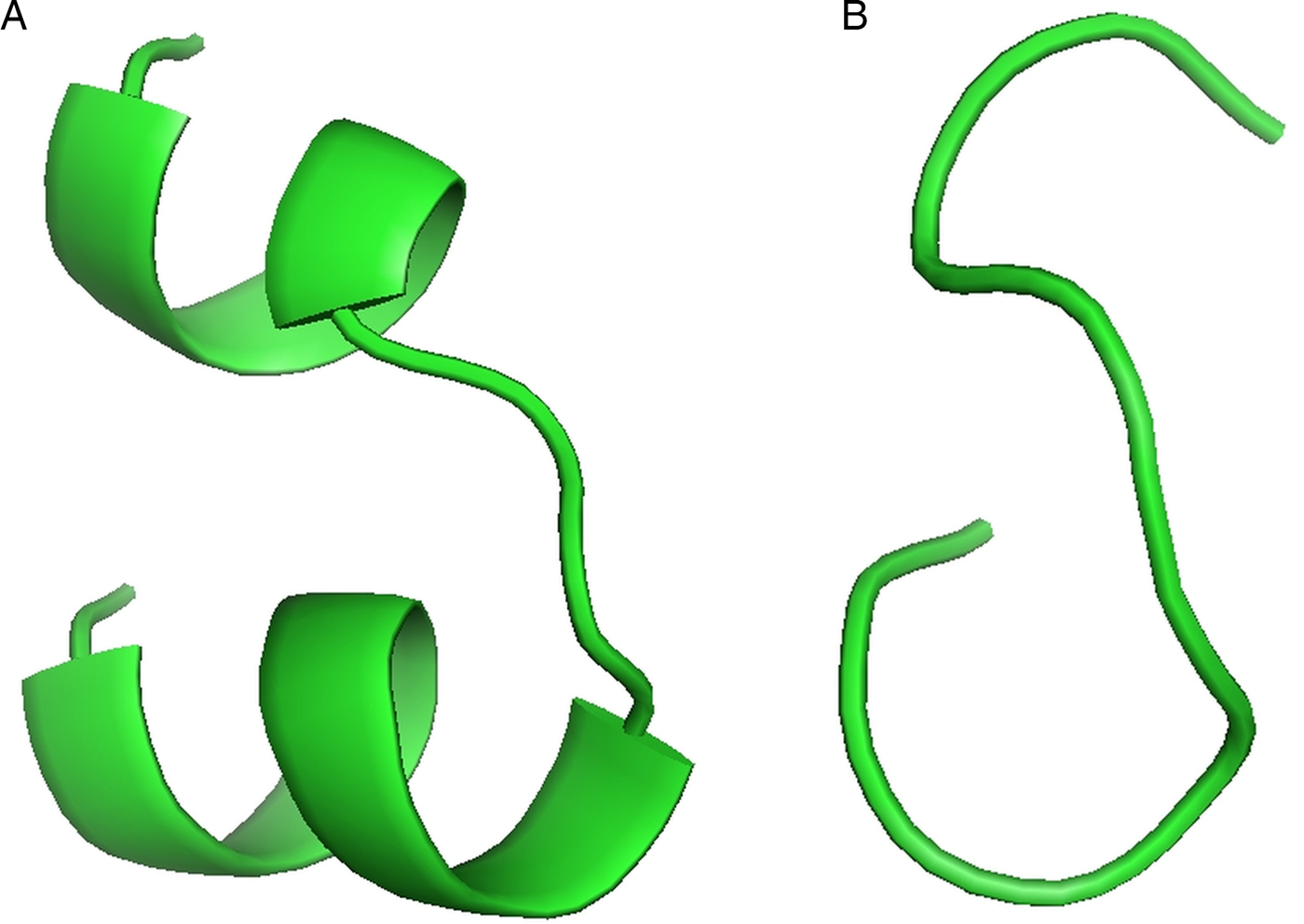
For the docking analysis and interaction with different HLAs, the top T-cell and B-cell predicted epitopes on cgd3_1400 (YANPGPIQY and HQMIETCIGFDSVTKS, respectively) were subjected to PEP-FOLD web-based server 3.5 for 3D structure conversion. This server modelled five 3D structures of the each proposed epitope, from which the best one was selected for the docking analysis (Fig. 2A and B).
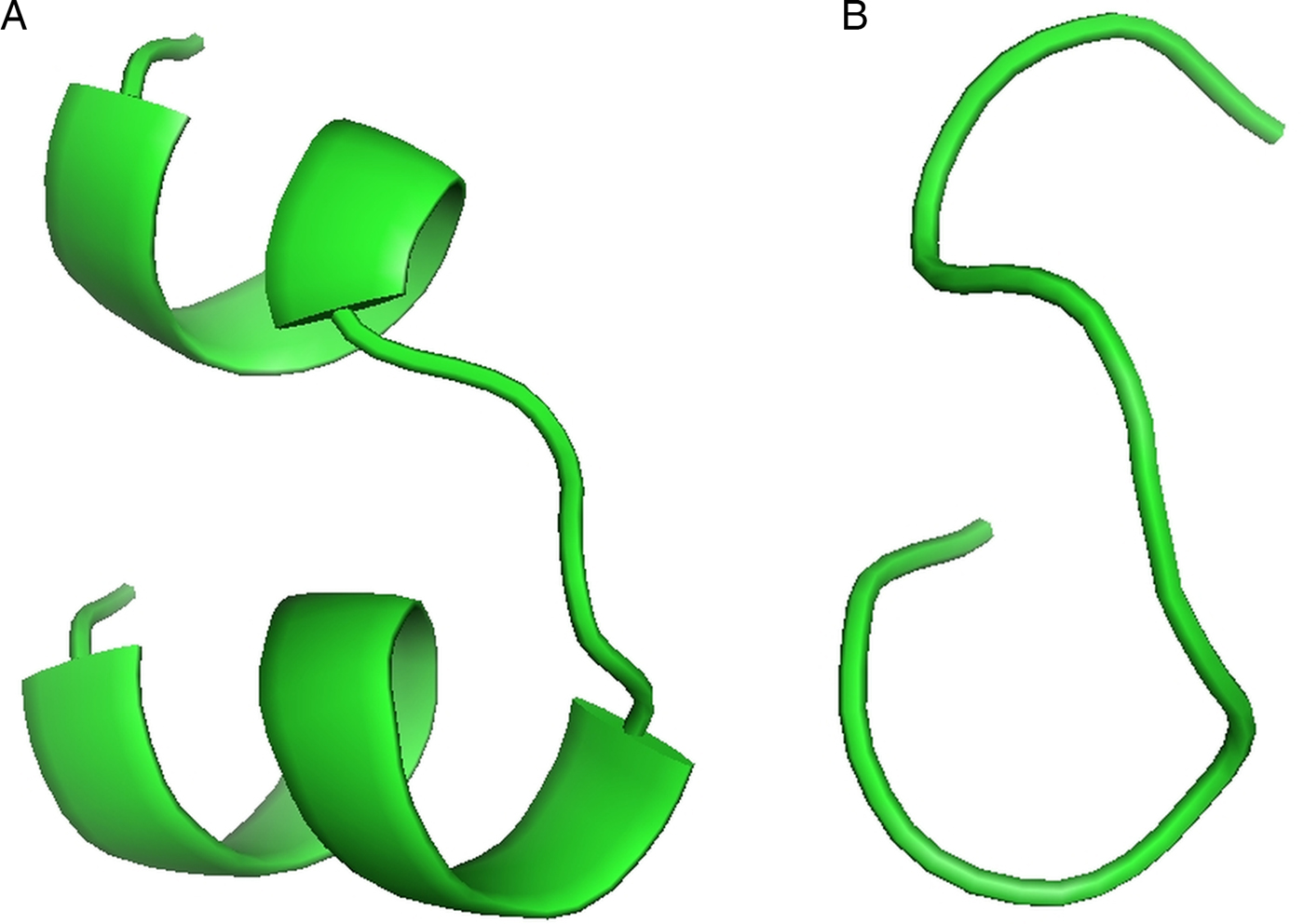
Fig. 2. (A) Predicted top scoring B-cell peptide structure by PEP-FOLD. (B) Predicted top scoring T-cell peptide structure by PEP-FOLD.
Using SWISS-MODEL workspace, the 3D structure modelling and optimization for cgd7_1830 (Secreted UDP-N-acetyl glucosamine pyro phosphorylase family protein, signal peptide) was performed with reference to template having PDB ID: 3oh0, after performing a BLASTP search against PDB database for the particular protein sequence (with 92% query coverage and 31% identity). Out of the three models predicted by the server, one is selected on basis of QMEAN and GMQE score (Fig. 3A). The predicted structure was submitted to SAVES server for quality check. Ramachandran plot generated by the PROCHECK server of the best model revealed that only 0.4% residues of the generated structure fall in the disallowed regions of the phi–psi dihedral angle plot, while 86.6% of the residues fall in the most favoured regions and 12.4% residues in the additional allowed regions, thus validating the quality of the model (Supplementary Fig. 1). The Verify3D program validated our selected model as having 87.75% of residues with an average 3D–1D score >0.2. The ERRAT server gives the overall quality factor (expressed as the percentage of the protein for which the calculated error values falls below the 95% rejection limit) of the model as 88.049.
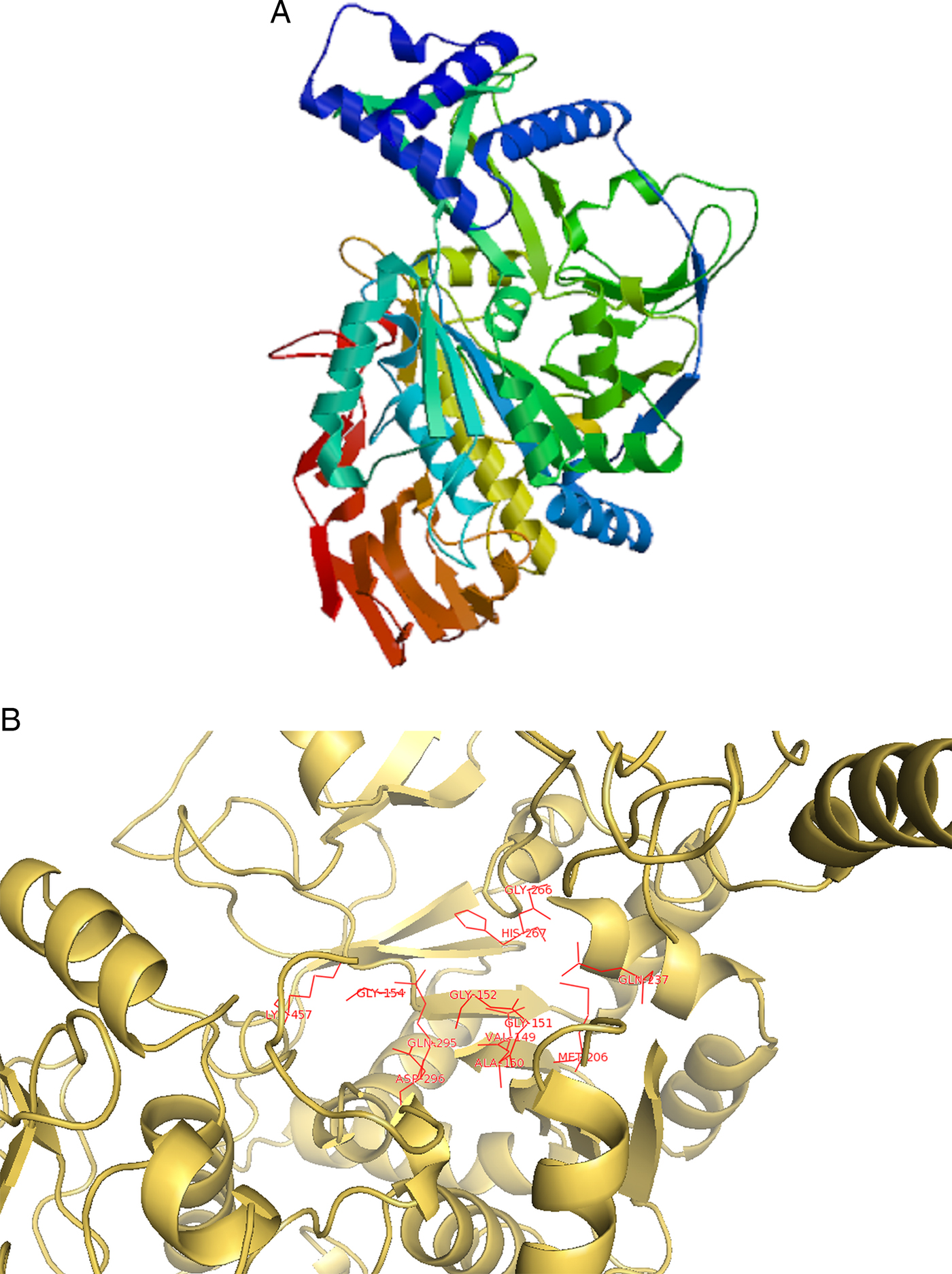
Fig. 3. (A) Predicted protein structure of cgd7_1830 by SWISS-MODEL. (B) Predicted active site residue of the protein predicted through 3DLigandSite on line server.
Docking analysis
The active site residues information of the SWISS-MODEL predicted protein structure of cgd7_1830 was inferred with the help of 3DLigandSite online server (Fig. 3B). The COFACTOR server was also used to infer a suitable ligand that will bind to the protein along with binding site residues. The result provided ligand ‘Uridine Tri Phosphate’ (UTP) with the highest C-score of 0.72. Several analogues of the lead ligand were retrieved from publicly available databases on the basis of similarity searches.
The ligands were divided into two sets, the first set consists of 371 compounds (after removing duplicates) comprising UTP and its similar compounds obtained from 3D similarity, literature and similar bioactivities search performed in PubChem database with respect to the lead ligand. This compound set also includes ligand obtained from 90% similarity search with the lead ligand in ChEMBL database and analogues obtained from the ZINC database. The second set of compounds consists of 11 anti-cryptosporidiosis compounds reported in Pathogen Box, which were downloaded and their file format was converted from smiles to sdf using OpenBabel software (O'Boyle et al. Reference O'Boyle2011). The anti-cryptosporidiosis compounds from Pathogen Box have successfully inhibited the pathogen growth in vitro conditions.
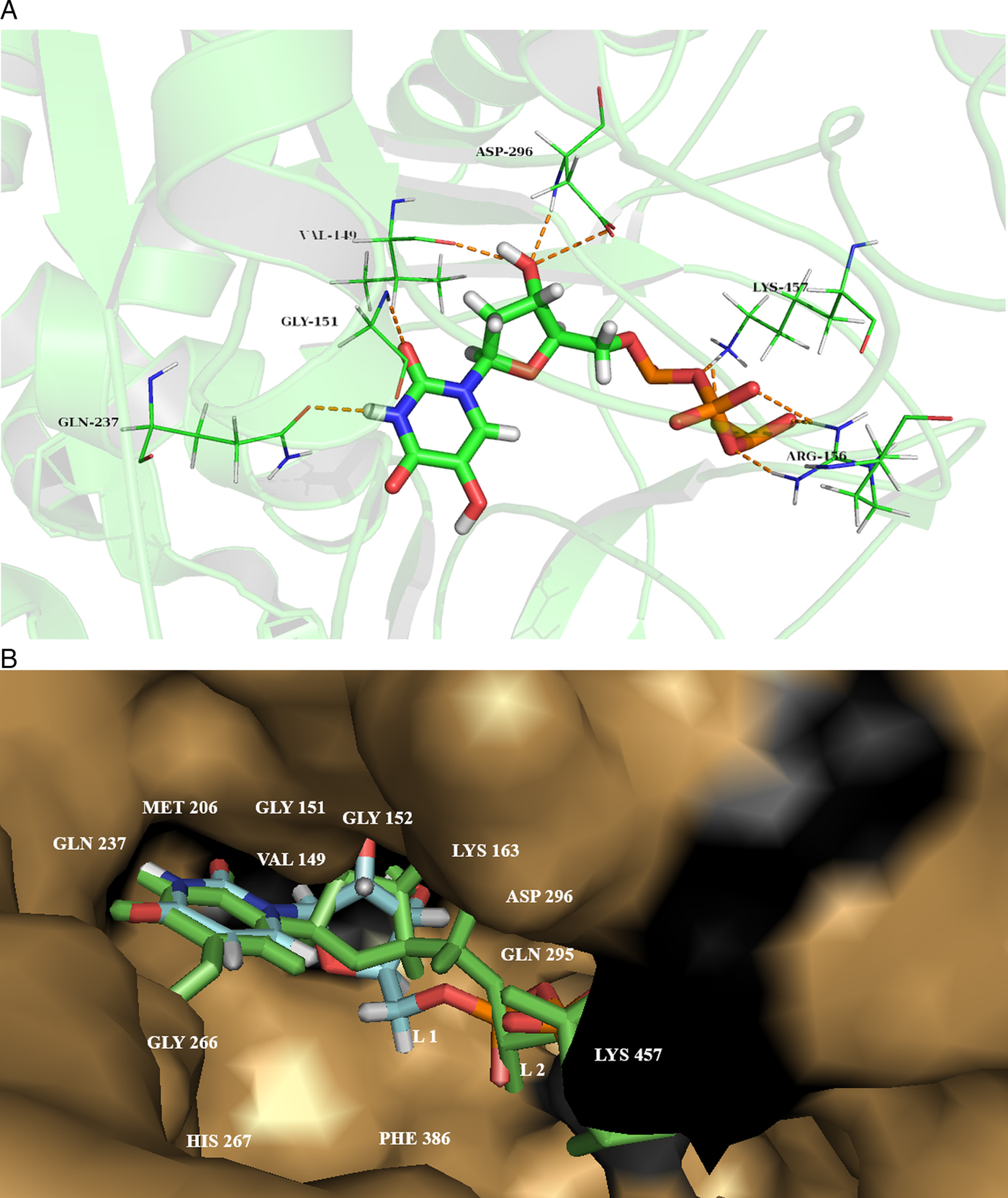
Later on, all 382 ligands were docked to our predicted drug target protein cgd7_1830 using Lead Finder implemented in Flare software module of Cresset®. Ligands were energy minimized with a gradient cut off of 0.200 kcal mol−1 A−1 with maximum 2000 iterations. Docking studies of all compounds were performed near the predicted active site residues. The ligands were ranked as per their binding energy. The compound having PubChem ID- 57371104 (hydroxy-[hydroxy-[[(2R, 3S, 5R)-3-hydroxy-5-(5-hydroxy-2, 4-dioxopyrimidin-1-yl) oxolan-2-yl] methoxy oxophosphaniumyl] oxyphosphoryl] oxy-oxophosphanium), reported the highest ‘Rank Score’ of −18.967 kcal mol−1 (Fig. 4A). The superimposed docking poses of the best hit along with the ligand ‘Uridine Tri Phosphate’ (UTP) docked conformation in the active sites of the predicted drug target protein is reported in Fig.4B. The docking scores of the top 10 compounds from various databases were provided in Supplementary Table 9.
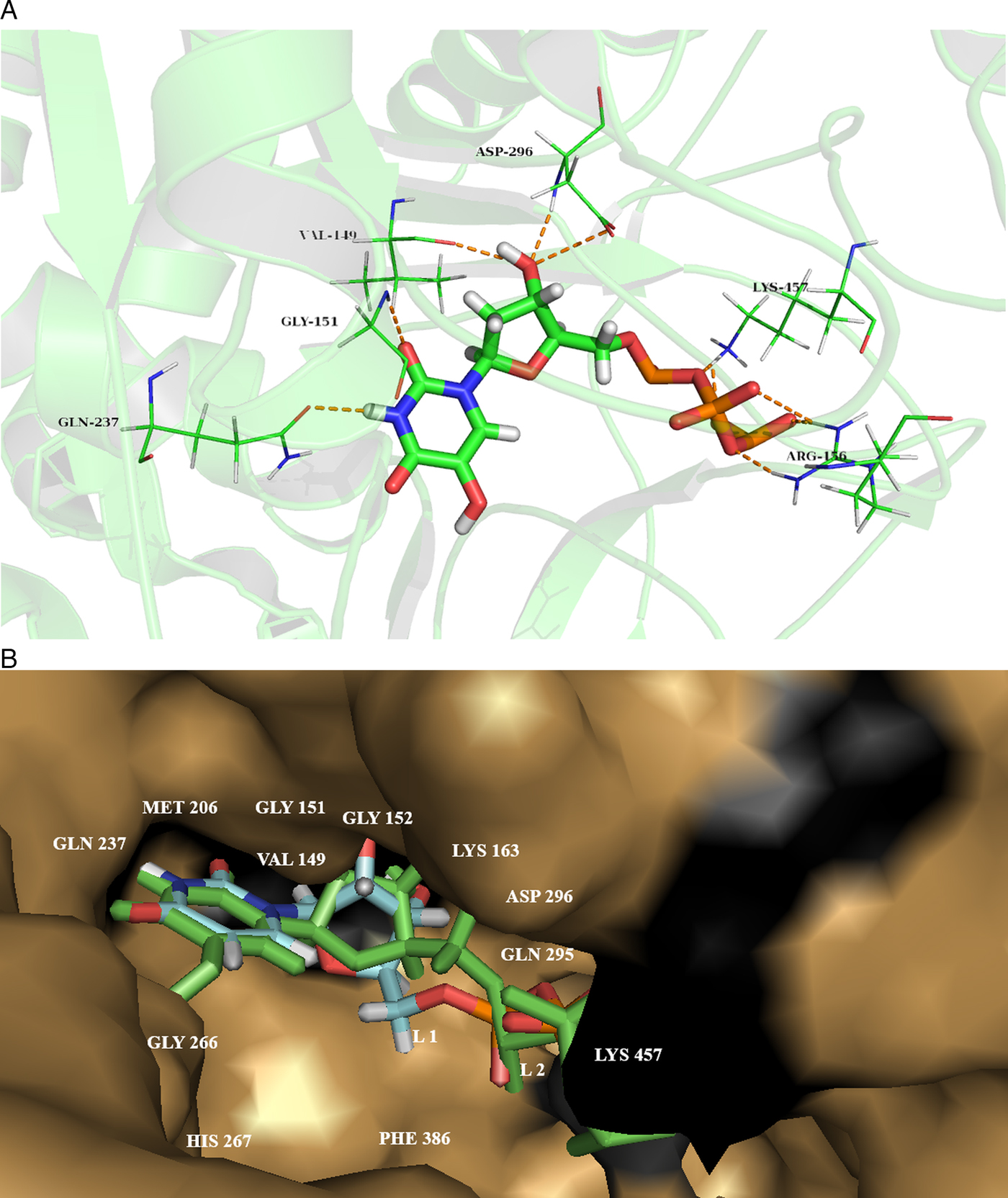
Fig. 4. (A) Compound that possesses highest binding score from the first compound set, PubChem ID 57371104 (hydroxy-[hydroxyl-[[(2R, 3S, 5R)-3-hydroxy-5-(5-hydroxy-2, 4-dioxopyrimidin-1-yl) oxolan-2-yl] methoxy oxophosphaniumyl] oxyphosphoryl] oxy-oxophosphanium). Hydrogen bonds (green dashed lines) are formed with residues VAL 149, GLY 151, GLN 237, HIS 267, ASP 296 and LYS 457. (B) Combine docking conformation of compounds; L1: ZINC000003861755 or UTP as default stick display and L2: PubChem ID- 57371104 as green stick display and within the active site pocket confirmation.
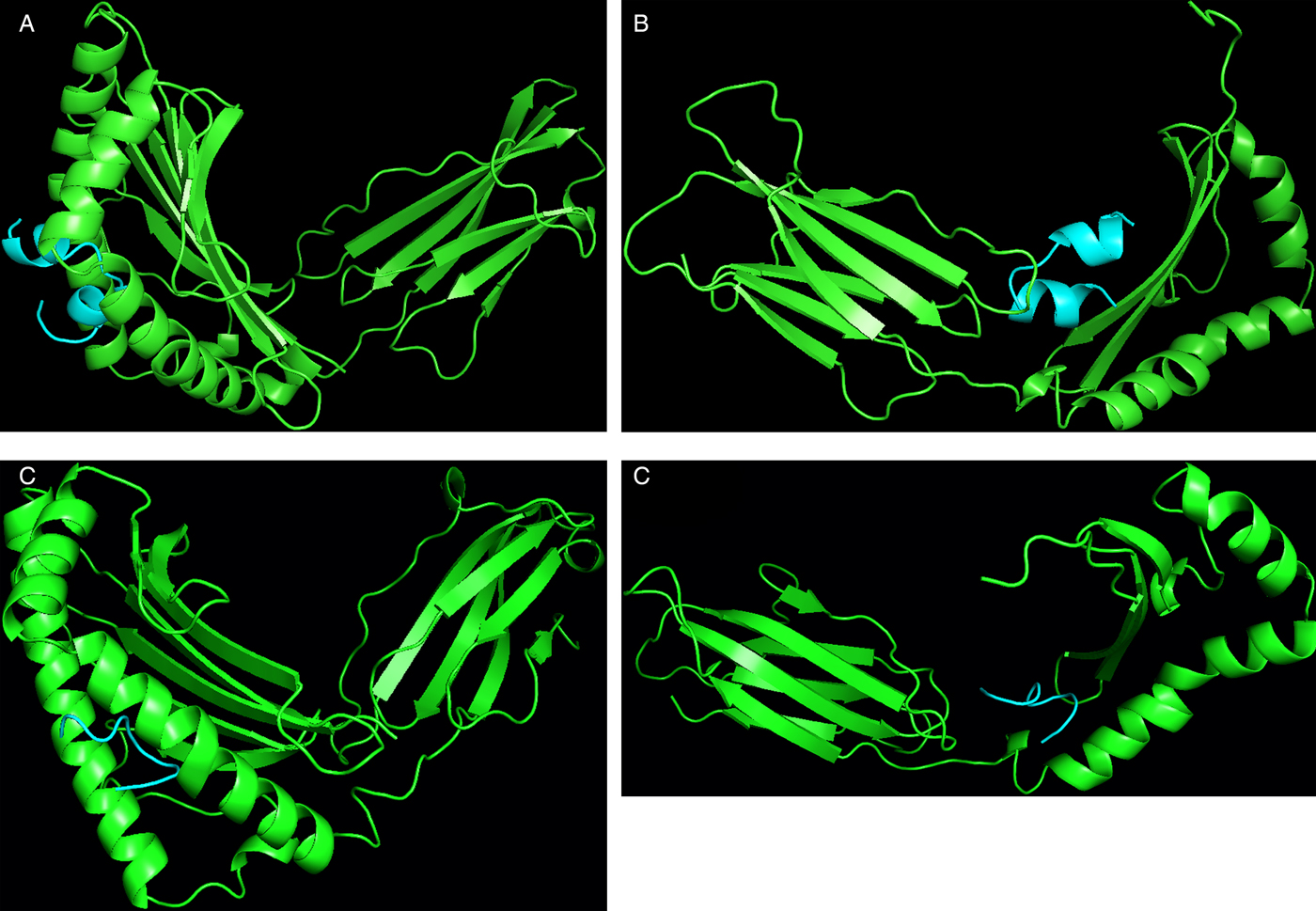
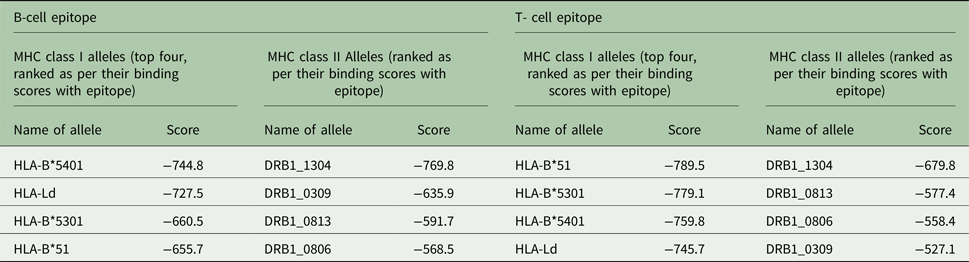
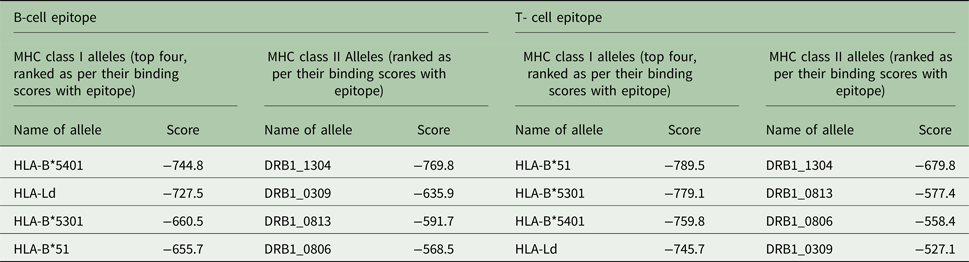
Similarly for docking of B- and T-cell epitopes with MHC class I and II alleles, at first the allele sequences were retrieved from UniProt database, NCBI-UniGene and some were retrieved from the ftpdirectory site of EMBL-EBI (European Bioinformatics Institute)’s IPD-IMGT/HLA datasets (Robinson et al. Reference Robinson2015). The top-ranking alleles as per their binding scores as predicted by ProPred and ProPred1 were subjected to docking with the predicted 3D structures of T and B-cell epitopes by PEP-FOLD individually in ClusPro server as shown in Fig. 5A–D. For the B-cell epitope, the highest binding score was reported with MHC class II allele DRB1_1304 and for the T-cell epitope, the highest binding score was reported with MHC class I allele HLA- B*51. The alleles were ranked as per their docking scores with the epitope structures (Table 2).
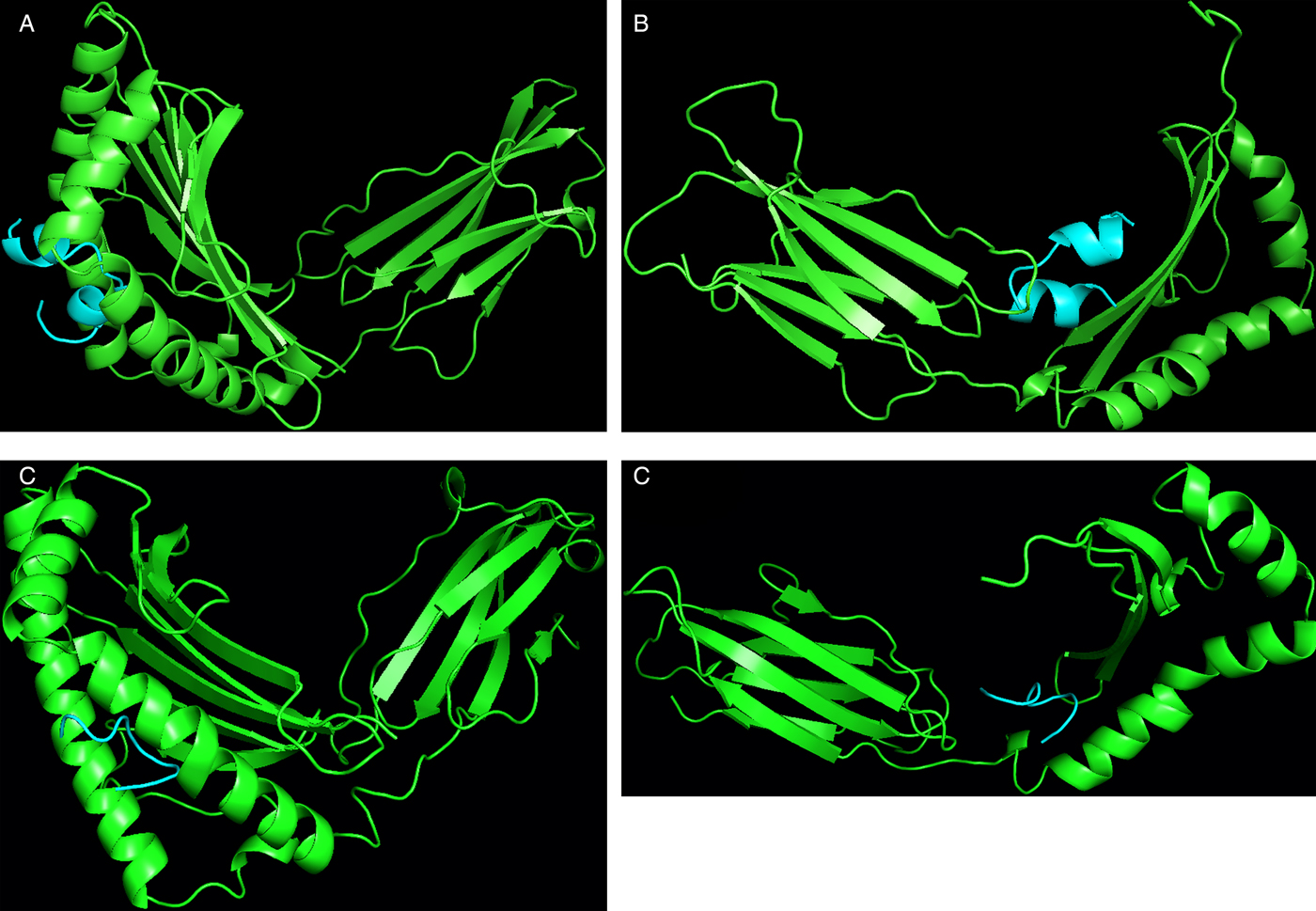
Fig. 5. Docking results of epitope peptides & MHC protein using ClusPro web server. (A) Docking of B-cell epitope with MHC-class I allele HLA- B*5401. (B) Docking of B-cell epitope with MHC class II allele DRB1_1304. (C) Docking of T-cell epitope with MHC class I allele HLA- B*51. (D) Docking of T- cell epitope with MHC class II allele DRB1_1304.
Table 2. MHC class I and II alleles ranked as per their docking scores with B and T-cell epitopes of cgd3_1400
Discussion
Cryptosporidiosis continues to be the major cause of diarrhoea in children particularly in the developing countries of Africa and Asia. Further, lack of a continuous in vitro culture system for Cryptosporidium proves to be the major impediments for experimental study against the parasite. In this regard, the computational study can be regarded as a cost-effective approach for identification of novel drug tagets and vaccine candidate.
In this study, different filtering approaches and screening processes have been used to filter the 103 proteins initially collected from different genomic databases to 11 non-homologous proteins, and then to seven essential proteins, which belong to the unique pathways of the pathogen, they are suitably chosen as useful drug and vaccine targets against the parasite. In our analysis, we identified proteins present in more than one pathway from the final list of seven proteins (Table 1). These can be regarded as excellent targets as blocking the enzyme activity will inhibit several metabolic pathways of the pathogen.
The proposed drug target protein cgd7_1830 (UniProtKB entry Q5CYM0) with a molecular weight of 72 915.79 Da, is of 654 amino acids in length and belongs to a UDPGP (UDP-glucose pyrophosphorylase) family of proteins (InterPro database ID: IPR002618), which catalyses the transfer of an uridylyl group. From KEGG database it is reported that an enzyme UDP-sugar pyrophosphorylase (USP)/UTP–monosaccharide-1-phosphate uridylyltransferase having EC number 2.7.7.64 is associated with this protein. USP enzyme acts on UTP and monosaccharide 1-phosphate to give a diphosphate and UDP–monosaccharide as products. This enzyme is involved in the formation of UDP-galactose in the galactose metabolism, D-glucuronate in ascorbate and aldarate metabolism and pentose and glucuronate interconversion, UDP-L-arabinose, UDP-glucose and UDP-galactose formation in amino sugar and nucleotide sugar metabolism and UDP-D-Xylose in Glucuronate pathway (uronate pathway). This reaction can be reversible in nature requiring Mg2+ or Mn2+ as catalysts for maximal activity and the enzyme shows broad substrate specificity towards monosaccharide 1-phosphates. However, mannose 1-phosphate, L-fucose 1-phosphate and glucose 6-phosphate are not substrates and UTP cannot be replaced by other nucleotide triphosphates (Kotake et al. Reference Kotake2004). Though it catalyses a monosaccharide 1-phosphate similar to UTP-glucose-1-phosphate uridylyltransferases (UGP2) (EC: 2.7.7.9), both are phylogenomically different and are orthologues. Moreover, it is also reported that UGP2 is not found in Cpv and USP is not found in human. USP has also been reported in some species of green alga Chlamydomonas and Chlorella, Aradopsis (Gronwald et al. Reference Gronwald, Miller and Vance2008), fungus Phytophthora infestans, Plasmodium berghei (as hypothetical protein), C. muris, Leishmania donovani, Toxoplasma gondii etc., but is absent from animals.
The proposed vaccine candidate protein cgd3_1400 (UniProtKB entry Q5CUU9) is of 148 083.40 Da and is 1327 amino acids in length. It is associated with many important pathways of the organism viz. glycolysis/gluconeogenesis, pentose phosphate pathway, fructose and mannose metabolism, biosynthesis of secondary metabolites and biosynthesis of antibiotics. The protein belongs to a family of pyrophosphate-dependent phosphofructokinase PfpB proteins (InterPro database ID IPR011183), which involves the transfer of phosphorus-containing groups. From KEGG database it is reported that enzyme pyrophosphate-dependent phosphofructo-1-kinase (PPi-PFK) having EC number 2.7.1.90 is associated with the protein. This enzyme acts on diphosphate and D-fructose 6-phosphate to give phosphate and D-fructose 1, 6-bisphosphate as products, which is the first committing step of glycolysis. Use of PPi as phosphate donor renders the reaction reversible, and can thus function both in glycolysis and gluconeogenesis. Though the enzyme catalyses 6-phosphofructokinase similar to ATP-dependent phosphofructokinase (ATP-PFK) (EC: 2.7.1.11) found in humans but utilizes diphosphate (PPi) as phosphoryl donor instead of ATP. PPi-PFK has been described in higher plants, primitive eukaryotes, bacteria and archaea, but is absent from animals (Carlisle et al. Reference Carlisle1990).
Another essential protein of the pathogen, cgd2_2130 (UniProtKB entry Q5CTQ1) also belongs to a family of pyrophosphate-dependent phosphofructokinase PfpB proteins (InterPro database ID IPR011183) having similar functions as that of cgd3_1400 and is also associated with the same enzyme PPi-PFK. For the essential plasma membrane protein, cgd3_2940 (UniProtKB entry Q5CUG3) no protein families or homologous super-families were found by an InterPro search. However, it contains a phospholipase D-like domains and is reported to be associated with transferring phosphorus-containing groups (EC: 2.7.8.-). From KEGG database, it is reported that the protein is involved in the formation of cardiolipin and glycerol from phosphatidylglycerol (Tan et al. Reference Tan2012). Due to unavailability of experimental data, though it was found that, the involved pathway is found in human, yet the involved enzyme is supposed to be different from that of a human. Other essential protein cgd5_70 (UniProtKB entry Q5CS64) belongs to Phosphoenolpyruvate carboxylase protein family (InterPro ID: IPR021135) and is reported to be associated with phosphoenolpyruvate carboxylase (PEPCase) enzyme (EC: 4.1.1.31). This enzyme is a key in the formation of oxaloacetate and orthophosphate in the tricarboxylic acid (TCA) cycle by carboxylation of phosphoenolpyruvate (Kai et al. Reference Kai, Matsumura and Izui2003). This enzyme plays an important role in carbon metabolism and pyruvate metabolism of Cpv, whereas absent in human.
Protein cgd8_380 (UniProtKB entry Q5CPY0) is found to be associated with malate: quinone oxidoreductase (MQO) (EC: 1.1.5.4) and belongs to FAD/NAD(P)-binding domain superfamily (InterPro ID: IPR036188). This enzyme is involved in the conversion of S-malate to oxaloacetate, with the reduction of a quinone (Kather et al. Reference Kather2000). This is a key reaction in TCA cycle and pyruvate metabolism. This enzyme is absent in humans. Protein cgd8_4940 (UniProtKB entry Q5CV93) is a Trehalose-phosphatase family protein as predicted by InterPro database (IPR003337) and is associated with two enzymes trehalose 6-phosphate synthase (EC: 2.4.1.15) and trehalose 6-phosphatase (EC: 3.1.3.12). Trehalose 6-phosphate synthase is a phosphoric-monoester hydrolase, which catalyses the de-phosphorylation of trehalose-6-phosphate to trehalose and orthophosphate; whereas, trehalose 6-phosphatase is a glycosyltransferase, which acts on UDP-alpha-D-glucose and D-glucose 6-phosphate to give UDP and alpha,alpha-trehalose 6-phosphate as products, which is an important step in starch and sucrose metabolism and trehalose biosynthesis (Pan et al. Reference Pan, Carroll and Elbein2002). It is reported that both the enzymes are not found in humans.
An epitope is a part of the antigenic molecule which can be recognized by T cells, B cells and antibodies (Kindt et al. Reference Kindt and Goldsby2007). Development of peptide vaccine is based on the fact that, only synthesizing the identified epitopes are sufficient to induce responses in the immune system. Structure modelling for the drug target protein and the predicted epitopes was performed. Finally, virtual screening and molecular docking studies were performed for the proposed drug and vaccine candidates.
Concluding remarks
Due to less availability of vaccine candidates and drug targets, cryptosporidiosis is emerging as the next threat in public healthcare domain. In our present study, a schema has been developed for drug discovery and vaccine candidate selection through computational methods using comparative genomics study.
Non-homologous and essential proteins are proposed to be essential drug targets and vaccine candidates. We selected two proteins (cgd3_1400 protein as vaccine candidate and cgd7_1830 protein as drug target), primarily on basis of their sub-cellular localization and other factors. For the possible vaccine candidate, we predicted B- and T-cell epitopes on cgd3_1400 protein, which will bind to MHC molecules. We determined the structures for both the top-ranking epitopes and drug target protein. Afterwards possible ligands for cgd7_1830 were screened through computational approaches. Cgd7_1830 protein is docked with the ligands and the epitopes were docked with MHC sequences obtained from various databases. Results from our study could facilitate the selection of proteins and possible ligands for entry into drug design production pipelines in the future.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182018000677
Acknowledgement
The authors would like to acknowledge the Bioinformatics laboratory facility of School of Biotechnology, KIIT University, during the course of the work.
Financial support
The project work is not supported by any external funding agency.
Conflicts of interest
None.
Ethical standards
Compliance with ethical standards.