


INTRODUCTION
Chagas disease caused by the protozoan parasite Trypanosoma cruzi is a major health problem in Latin America. The most frequent and severe clinical consequence of the infection is chronic Chagas' Heart Disease (cChHD), affecting approximately 30% of the infected people. Cardiac manifestations include atrio- and intraventricular conduction disturbances, cardiomegaly and sustained brady- and tachyarrthythmias that may lead to sudden death (Elizari and Chiale, Reference Elizari and Chiale1993; World Health Organization, 2000; Halperin and Rassi, Reference Halperin and Rassi2000). Experimental results and analysis of the human antibody response against the parasite ribosomal P proteins have suggested a link between high antibody levels to these antigens and the induction of arrhythmias (De Oliveira et al. Reference De Oliveira, Pedrosa, Nascimento, Campos de Carvalho and Masuda1997; Kaplan et al. Reference Kaplan, Ferrari, Lopez-Bergami, Mahler, Levitus, Chiale, Hoebeke, Van Regenmortel and Levin1997; Lopez Bergami et al. Reference Lopez Bergami, Scaglione and Levin2001). The main epitope of the T. cruzi ribosomal P2β protein (TcP2β) in this chronic infection was mapped to the C-terminus of this antigen, a highly negatively charged stretch of 13 amino acid residues (peptide R13, EEEDDDMGFGLFD) (Sepulveda et al. Reference Sepulveda, Liegeard, Wallukat, Levin and Hontebeyrie2000; Lopez Bergami et al. Reference Lopez Bergami, Gomez, Levy, Grippo, Baldi and Levin2005). Interestingly, the R13 peptide differs only in 1 amino acid with the C-terminal region (peptide H13, EESDDDMGFGLFD) of the human ribosomal P proteins, which is in turn the target of anti-P autoantibodies in Systemic Lupus Erythematosus (SLE). These facts led several authors to postulate that the underlying mechanisms of the anti-P autoantibodies may be similar in Chagas and SLE diseases (Skeiky et al. Reference Skeiky, Benson, Parsons, Elkon and Reed1992, Reference Skeiky, Benson, Guderian, Sleath, Parsons and Reed1993). Further studies with polyclonal mono-specific anti-P antibodies showed that autoantibodies developed in SLE patients reacted with human and parasite P proteins to the same extent, while anti-P antibodies from cChHD patients showed a marked preference for the parasite proteins (Kaplan et al. Reference Kaplan, Ferrari, Lopez-Bergami, Mahler, Levitus, Chiale, Hoebeke, Van Regenmortel and Levin1997). Human recombinant antibodies would be indeed excellent tools to elucidate the role and fine specificity of these anti-P antibodies in cChHD.
The selection of antibodies from combinatorial libraries displayed on the surface of filamentous phage has become important for the generation of reagent, diagnostic, and therapeutic molecules and for the study of the natural immune response (Hoogenboom, Reference Hoogenboom2005; Thie et al. Reference Thie, Meyer, Schirrmann, Hust and Dübel2008; Dübel et al. Reference Dübel, Stoevesandt, Taussig and Hust2010). Phage display of combinatorial libraries has been used to obtain recombinant human antibodies from patients infected with Plasmodium falciparum (Roeffen et al. Reference Roeffen, Raats, Teelen, Hoet, Eling, Van Venrooij and Sauerwein2001; Lundquist et al. Reference Lundquist, Nielsen, Jafarshad, Soesoe, Christensen, Druilhe and Dziegiel2006), or Plasmodium yoelii (Vukovic et al. Reference Vukovic, Chen, Qin Liu, Foley, Boyd, Kaslow and Good2002), or from mice infected with Cryptosporidium parvum (Chen et al. Reference Chen, Williams, Yang, Cevallos, Bhat, Ward and Sharon2003) or Toxoplasma gondii (Hoe et al. Reference Hoe, Wan and Nathan2005). Although no human mAbs against any T. cruzi protein from cChHD patients have been fully characterized up to date.
To better understand the molecular mechanisms of epitope-paratope interactions that seem to be related to the pathogenesis of human heart disease in chronic T. cruzi infections, we cloned human single-chain variable fragment (scFv) antibodies to the TcP2β and compared them to autoanti-P recombinant antibodies obtained from SLE patients. Our results confirmed the very specific nature of the anti-T. cruzi ribosomal P protein response to the ribosomal P2β protein in cChHD, and also demonstrated that construction of human scFv libraries accompanied by an appropriate panning strategy, may be used to obtain recombinant human anti- T. cruzi mAbs in a relatively short time.
MATERIALS AND METHODS
Cloning, expression and purification of recombinant proteins
The TcP2β gene was cloned into pMalc2 (New England Biolabs, Cambridge, MA, USA) and pGex-1λT (Pharmacia Biotech, Uppsala, Sweden) vectors in the EcoRI site. Production and purification of the maltose-binding protein (MBP) and gluthatione-S-transferase (GST) fusion proteins, MBP-TcP2β and GST-TcP2β, were performed as indicated by the manufacturers.
Synthetic peptides
Peptides were prepared by the solid-phase method of Merrifield as described by Müller et al. (Reference Muller, Couppez, Briand, Gordon, Sautiere and Van Regenmortel1985) with a semi-automatic multi-synthesizer NPS 4000 (Neosystem, Strasbourg, France). Peptide R13 (EEEDDDMGFGLFD) was derived from the 13 carboxyl-terminal amino acids of TcP2β (Levin et al. Reference Levin, Vazquez, Kaplan and Schijman1993), while peptide H13 (EESDDDMGFGLFD) corresponds to the C-terminal region of the mammalian ribosomal P proteins (Elkon et al. Reference Elkon, Bonfa, Llovet, Danho, Weissbach and Brot1988). Peptide TMVP (AEAALUKMALMKV) was derived from tobacco mosaic virus coat protein. Peptides were coupled at a molar ratio of 1:30 to bovine serum albumin (BSA) (Sigma, St Louis, MO, USA) with 0·05% glutaraldehyde as described (Müller et al. Reference Muller, Couppez, Briand, Gordon, Sautiere and Van Regenmortel1985).
Patients
Fourteen cChHD patients were evaluated at the Cardiology Unit of the Hospital Fernández (Buenos Aires, Argentina) and underwent a complete clinical and cardiological examination. Ventricular arrythmias and sinus node dysfunction were diagnosed as reported previously by Chiale et al. (Reference Chiale, Ferrari, Mahler, Vallazza, Elizari, Rosenbaum and Levin2001). After clinical evaluation, blood samples were drawn, and sera were obtained and coded. ELISA and Western blots against T. cruzi epimastigote lysate and several T. cruzi antigens as peptide R13 were performed. According to these results, patient 5 (one of the patients with the highest titres of anti-T. cruzi and anti-TcP2β antibodies) was selected to be the bone-marrow donor to construct the scFv library. Clinical features of this patient are shown in Table 1.
Table 1. Clinical features of patient 5 (bone-marrow donor)

The study protocol complied with the Helsinki Declaration and was approved by the Committee for Ethical and Legal aspects of Research (CELAR) of the Institute for Genetic Engineering and Molecular Biology, Buenos Aires, Argentina.
Preparation of RNA from bone marrow and cDNA synthesis
By sternum puncture of patient 5, 15 ml of bone marrow were obtained and immediately added to a 50 ml tube containing denaturing solution (DS=8·4 m guanidine thiocyanate, 0·7% trisodium citrate and 0·5% sarcosyl in DEPC-treated water, adding immediately 100 μl of β-mercaptoethanol to each 14 ml of DS before use). To each 10 ml of the sample (bone marrow+DS), 1 ml of 3 m sodium acetate (pH 4), 10 ml of acidic phenol and 2 ml of chloroform: isoamilic alcohol (24:1) were added and the mixture was very well vortexed. After 15 min chilling on ice, the samples were centrifuged for 30 min at 10 000 g, and the supernatants were carefully aspirated and put into clean RNAse-free Corex® tubes. One volume of isopropanol was added and left 10 min at room temperature (RT). After a 20 min centrifugation at 10 000 g and washing with 75% ethanol, the pellet was resuspended in 200 μl of RNAse-free water. The RNA concentration was quantified with a UV-spectrometer (277 μg of total RNA were obtained) and its quality was assessed on an agarose gel, confirming the integrity of rRNA. To obtain cDNA, first strand synthesis using reverse transcriptase standard procedure (Life Technologies, CA, USA) was performed according to the manufacturers’ instructions. The cDNA was UV-quantified and stored at −20°C.
ScFv library construction
The cDNA was used as a template for amplification of the human IgG variable heavy (VH) and variable light (VL) chain gene fragments. VH gene regions were amplified using a combination of 6 forward and 1 reverse primers, respectively; while the V kappa (Vκ) gene regions were amplified using 4 forward and 4 reverse primers and V lambda (Vλ) gene regions were amplified using a set of 9 forward and 3 reverse primers (Andris-Widhopf et al. Reference Andris-Widhopf, Steinberger, Fuller, Rader, Barbas, Barbas, Burton, Scott and Silverman2000). For each V gene amplification, a total reaction volume of 100 μl containing 0·5 μg template cDNA, 60 pmoles of each forward and reverse primer, 1× PCR buffer, 2 mm MgCl2, 200 mm dNTP (Promega, USA) and 2·5 U Taq polymerase (Life Technologies, USA) was used.
Two reactions were performed for each Vλ and Vκ amplification and 5 reactions were performed for each VH amplification. The amplifications were performed in a Perkin Elmer (MA, USA) Thermocycler with the following programme: 94°C for 5 min, followed by 30 cycles of 94°C for 15 sec, 56°C for 15 sec and 72°C for 90 sec, and concluded with a final elongation step at 72°C for 10 min. The corresponding amplified products were purified using the Qiaquick Gel Extraction Kit (Qiagen, Germany). Subsequently, the purified VL and VH chain fragment coding sequences were fused in a second PCR using the primers RSC-F and RSC-B (Andris-Widhopf et al. Reference Andris-Widhopf, Steinberger, Fuller, Rader, Barbas, Barbas, Burton, Scott and Silverman2000) to produce the complete scFv fragment. For this procedure, 120 reactions of 100 μl were prepared and each of these contained 0·1 μg of purified VH and VL genes respectively, 60 pmoles of each RSC-F and RSC-B, 200 mm dNTP, 1× PCR buffer, 2·5 U Taq polymerase (Life Technologies, USA) and sterile deionized water. The PCR reactions were performed with the following programme: 94°C for 1 min, followed by 30 cycles of 94°C for 15 sec, 56°C for 15 sec and 72°C for 2 min. ScFv fragments were gel purified and restricted with SfiI (16 U/μg DNA, 6 h at 50°C). This mixture was subjected to gel purification and used in a test ligation as follows: 700 ng of DNA were ligated into 560 ng of SfiI restricted pComb3X vector (Scripps Research Institute, USA) (gel purified) with 1 U of ligase (Life Technologies, USA) in a 10 μl reaction, overnight at 16°C. The ligated product was ethanol-precipitated and resuspended in 10 μl of sterile distilled water and transformed into electrocompetent Escherichia coli XL1-BlueF′ (New England Biolabs, USA) by electroporation (2·5 kV, 200 ohms, 25 mF). After the library size was estimated, 4 library ligations were performed by scaling up the reactions by a factor of 10 under similar conditions. For library ligation, 5·6 μg of vector were combined with 7 μg of digested scFv fragment. DNA was precipitated and transformed into 300 μl of electrocompetent cells. Eight electroporations were carried out.
Panning against T. cruzi ribosomal P2β protein (TcP2β) and its C-terminal end
Phage bearing scFv fragments on their surface were selected by panning on antigen-coated wells. Microtitre wells (Nunc Immunoplates, Roskilde, Denmark) were coated with 500 ng of MBP-TcP2β recombinant protein in 0·1 m NaHCO3, pH 9·6. For panning against peptide R13 (the C-terminal end of TcP2β), wells were incubated with 250 ng streptavidin for 2 h at 37°C followed by incubation for 1 h with 500 ng biotinylated R13 peptide and incubated overnight at 4°C . The wells were then blocked with 3% BSA PBS-0·1% Tween 20 (PBST) and polyethyleneglycol-precipitated phage were added (50 μl/well) and incubated for 2 h at 37°C. Unbound phage were washed with PBST 5 times (in the 3 subsequent panning rounds washing was increased up to 15 washes). Bound phage were eluted with glycine-HCl, pH 2·2 and then neutralized with 1 m Tris-HCl, pH 8. The eluted phage were then allowed to infect E. coli XL1-BlueF′cells to be amplified in the presence of the VCSM13 helper phage (Stratagene, USA). Amplified phage were submitted to 3 further rounds of panning. Input and output phage were titrated on LB-carbenicillin plates.
Phage-ELISA
To determine enrichment in specific phage bearing anti-TcP2β antibodies, the original unpanned library and the resultant phage pool (output) from each subsequent round of panning were analysed by phage ELISA. For this purpose, microtitre plate wells were coated with MBP-TcP2β or peptide R13 as described above and blocked with 3% BSA PBST. MBP was used as protein control. Phage preparations at a dilution of 1 in 2 were added (50 μl/well) and incubated for 2 h at 37°C. Each well was extensively washed 5 times with PBST and HRP-conjugated anti-M13 secondary antibody (Pharmacia, 1:3000) was added and incubated 1 h at 37°C. Enzyme activity was revealed with 100 μl of TMB substrate (3,3′,5,5′-tetramethylbenzidine dihydrocholoride (Sigma) at a concentration of 3 mg/ml in: 0·1 m citrate buffer pH 7; 2% DMSO; 1·8% glycerol; 0·01% H2O2) per well. The enzymatic reaction was stopped after 10 min by adding 50 μl of 1 m HCl and the OD at 450 nm was read with an automated plate reader (Molecular Devices, CA, USA).
For reactivity measurement of individual clones, amplified monoclonal phage were used instead of polyclonal phage preparations at a dilution of 1 in 2 as described above. Cut-off values corresponded to the mean plus 3 s.d. of the reactivity measured in control phage that did not recognize either TcP2β or peptide R13. Clones with OD values above the cut-off line were considered positive for respective antigens.
scFv-PCR and BstOI fingerprinting
Individual clones after library transformation and panning were picked and introduced into a PCR tube containing 10 μl of nuclease-free water. Forward primer ompseq (5′AAGACAGCTATCGCGATTGCAG3′) and reverse primer gback (5′GCCCCCTTATTAGCGTTTGCCATC3′) were used to amplify the scFv fragment. PCR products were separated on a 2% agarose gel. The amplified PCR products were digested with 15 U BstOI (Promega, USA) for 2 h at 60°C, and fingerprints were analysed on 4% agarose gels.
Recombinant scFv expression and periplasmic extraction
To produce soluble scFv, phagemid DNA from selected clones was transformed into TOP10 cells (Invitrogen, USA). Following an overnight incubation on LB-carbenicillin plates at 37°C, single colonies were inoculated into 3 ml of SB (Super Broth). Starters were inoculated into 100 ml culture 2×yeast/tryptone (2×YT) culture medium and growth at 37°C for 3 h (OD600 0·8–1·0), IPTG was added to a final concentration of 1 mm, followed by an overnight incubation at 20°C. The cultures were centrifuged and the cell pellet was resuspended in a solution of 5 mm Tris, 1 mm EDTA, 20% sucrose plus a protease inhibitor cocktail tablet (Roche, Germany) and subjected to shaking for 1 h at RT. After a centrifugation step, the supernatant fraction containing the scFv (periplasmic extract) was recovered and stored at 4°C until required.
ScFv C5 against TcP2β and peptide R13 was prepared as described by Smulski et al. (Reference Smulski, Labovsky, Levy, Hontebeyrie, Hoebeke and Levin2006). The anti-P scFv A4 and C10 derived from SLE patients were prepared as described by Zampieri et al. (Reference Zampieri, Mahler, Bluthner, Qiu, Malmegrim, Ghirardello, Doria, Van Venrooij and Raats2003).
scFv-ELISA
ELISA assays were performed as described by Mesri et al. (Reference Mesri, Levitus, Hontebeyrie-Joskowicz, Dighiero, Van Regenmortel and Levin1990). Briefly, 96-microwell plates were coated with 50 μl of 5 μ m BSA-conjugated peptides or with 50 μl of 5 μg/ml GST-TcP2β in 0·05 m bicarbonate–carbonate buffer (pH 9·6) and blocked for 1 h at 37°C with skim milk 5%-PBST. BSA and peptide TMVP were used as controls. Periplasmic scFv extract (see above for preparation) were used at a dilution of 1 in 2 and incubated for 2 h at RT. The reaction was visualized using a peroxidase-conjugated mouse secondary anti-histidine (anti-His-HRP) (Sigma, CA, USA) at a dilution of 1:5000 followed by TMB-H2O2 substrate. Reaction was stopped with 1 m HCl and colour development was monitored at 450 nm.
Western blotting
The presence of the scFv in the periplasmic extract was assayed by Western blotting (Towbin et al. Reference Towbin, Staehelin and Gordon1979). Expressed proteins (periplasmic extract) were separated on a 15% SDS-polyacrylamide gel and subsequently transferred to a nitrocellulose membrane. The membrane was blocked overnight with 3% skim milk in TBS-0·1% Tween 20 (TBST) at 4°C. The membrane was then incubated with an anti-His-HRP Ab (1:7500) for 1 h followed by 3×10 min wash cycles with TBST and subsequently, TMB-H2O2 substrate was added.
For the analysis of scFv specificity towards TcP2β, 5 μg of MBP-TcP2β was separated on a 12% SDS-polyacrylamide gel and transferred to a nitrocellulose membrane. MBP was loaded in the same well as control. The membrane was blocked overnight (3% milk in TBST), washed thoroughly and co-incubated with the perisplamic extract and the anti-His-HRP Ab (1:7500) for 2 h at RT, followed by 5×5 min washes with TBST and 1×5 min with TBS. The membrane was then revealed as described above.
Sequence analysis
Nucleic acid sequencing was carried out in our laboratory on a Megabace 500 (Molecular Dynamics, CA, USA) using template amplification with primers ompseq, gback (see above) and leadVH (5′GCTGCCCAACCAGCCATGGCC) for sequencing with an adaption of the Templiphi protocol (Amersham Biosciences, standard operating procedure/protocol by Amir Ghadiri/ production sequencing group, September 5, 2001). VH and VL sequences were analysed with IgBlast program (National Library of Medicine, NIH; www.ncbi.nlm.nihg.gov/blast/igblast) and the IMGT/V-QUEST alignment tool at IMGT, the international ImMunoGeneTics database (Lefranc, Reference Lefranc2003). Alignment analyses were conducted using MEGA version 4 (Tamura et al. Reference Tamura, Dudley, Nei and Kumar2007). Mutations were identified comparing each sequence with germline sequences and were defined on the basis of nucleotide changes in the VH or VL segment. Replacement (R) and silent (S) mutations were determined in framework regions (FRs) and complementary-determining regions (CDRs). The probability that an excess or scarcity of R mutations in VH CDR or FR occurred by chance was calculated by a multinomial distribution model (Lossos et al. Reference Lossos, Tibshirani, Narasimhan and Levy2000).
Statistical analysis
The results were statistically analysed with the paired Student's t-test, and P value <0·05 was considered significant. The P value, probability that excess (for CDR) or scarcity (for FR) of mutations occurred by chance, were calculated by multinomial distribution model (Lossos et al. Reference Lossos, Tibshirani, Narasimhan and Levy2000).
RESULTS
ScFv library construction
A human scFv library was generated from mRNA of a cChHD patient (patient 5) with a severe sinus node dysfunction. At the serological level, a high anti-T. cruzi antibody titre together with a strong reactivity to the parasite ribosomal P proteins and epitope R13 were measured (Table 1).
In order to obtain a major repository of antibody-secreting plasma cells (Burton, Reference Burton, Barbas, Burton, Scott and Silverman2000), total RNA from bone marrow was isolated (Fig. 1A). To amplify the variable regions of the transcribed antibodies, cDNA was generated by reverse transcription. Using several pairs of different primers variable heavy (VH) and light (VL) chain fragments were amplified. The VL forward primers and VH reverse primers were designed to incorporate asymmetric SfiI restriction sites to facilitate directional subcloning into pComb3X phagemid vector system (Andris-Widhopf et al. Reference Andris-Widhopf, Steinberger, Fuller, Rader, Barbas, Barbas, Burton, Scott and Silverman2000). In addition, the 3′ end of the amplified VL products and the 5′ end of the amplified VH products both had a short linker sequence of 7 amino acids (3 glycine and 4 serine residues) to connect VL and VH regions into an scFv fragment. The amplified VH and VL products were electrophoresed and the expected fragment sizes of 400 bp and 350 bp respectively were observed (Fig. 1B). The assembled scFv (750 bp) was obtained by overlap PCR (Fig. 1C).

Fig. 1. Library construction. (A) Bone marrow total RNA from the cChHD patient. (B) VH (1–6) and VL (7–50) amplifications from cDNA. Negative control (−) was without cDNA as template. Lane M, 1 kb plus DNA marker (Invitrogen). (C) scFv fragment obtained by overlap-PCR of VH and VL amplification products. (D) pComb3X-scFv construct. The scFv gene is directionally cloned into the phagemid vector using a unique SfiI cloning step between the leader sequence ompA and gene III (gIII). ScFv is expressed in a suppressor E. coli strain as fused protein scFv-gIII for phage display. The displayed protein has hexa-histidine (H6) and haemagglutinin (HA) peptide tags for scFv purification and detection. The vector also carries an amber stop codon (TAG) to allow the expression of soluble antibodies in a non-suppressor E. coli strain as TOP10F′.
The final PCR product was cloned into pComb3X vector, as illustrated in Fig. 1D, and the library was transformed into XL1-Blue F′ cells. The total scFv library contained 2×109 cfu per μg of transformed DNA. Library quality and diversity was analysed by PCR for full-length inserts and BstOI fingerprint restriction, respectively. Thus, 87 out of 94 clones (92·5%) had full-length inserts, and analysis of the BstOI fingerprint pattern indicated high diversity among them (data not shown).
Screening of specific anti-TcP2β clones
Recombinant phage expressing scFv as a fused protein with phage pIII coat protein were produced by helper phage rescue and panned against TcP2β and peptide R13. After the initial round of panning, the output number resulted in a low enrichment value of 0·14 in round 2 for TcP2β probably due to the high stringency in washing conditions (data not shown). However, an enrichment of almost 17-fold was observed for R13 peptide. After round 3, there was a 12·7-fold increment in the number of specific phage for TcP2β but an increase of only 7·29-fold for peptide R13. The enrichment value increased to 23·2-fold for TcP2β and 308·4-fold for peptide R13 after the final panning round, indicating enriched specific binding to each antigen.
Phage ELISA was performed to detect the presence of enriched phage carrying specific scFv towards TcP2β and peptide R13 over 4 rounds of panning (Fig. 2A). Enrichment of specific scFv-bearing phage during selection was from an OD value of 0·2 at the first round up to 2·3 at the fourth round for TcP2β and from 0·11 up to 1·8 for R13 peptide. The absorption value of the unselected library was similar to helper phage (VCSM13) control. The specificity of the interaction was evident by the low binding of the selected phage to MBP (Fig. 2A).
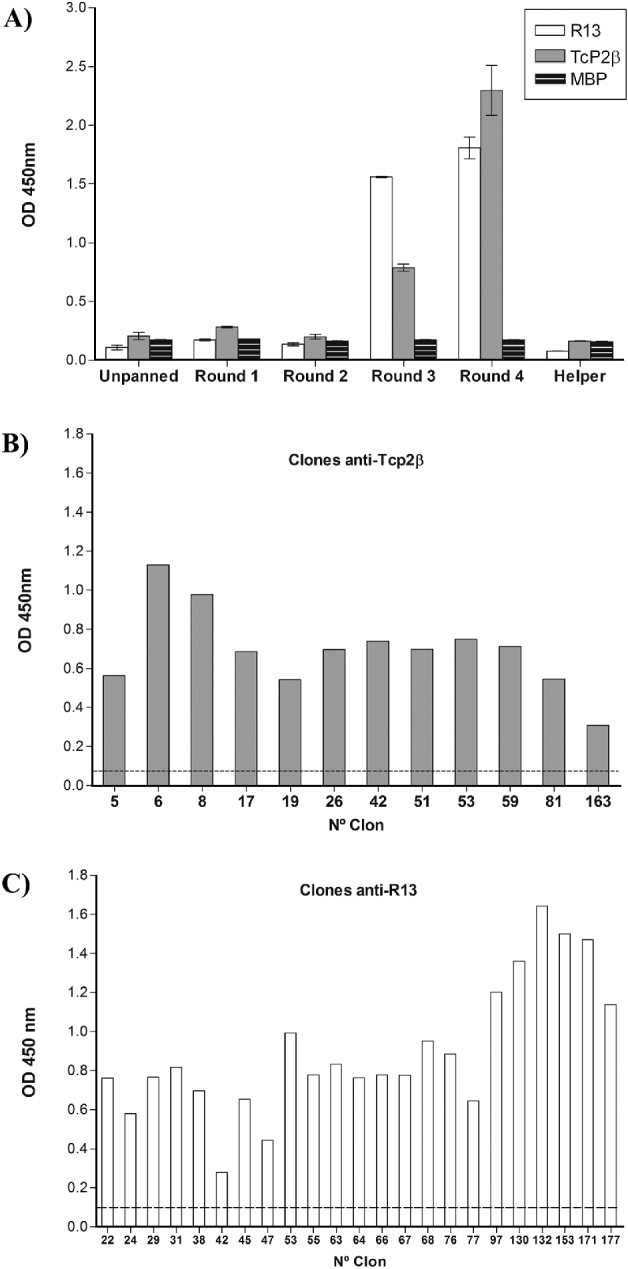
Fig. 2. Identification of reactive phage against TcP2β derived from an scFv library by Phage ELISA. (A) Reactivity of unpanned library and output phage after each round of panning against TcP2β and its C-terminal end (R13 peptide). Helper phage VCSM13 was used as control as well as the non-related MBP protein. (B) Reactivity against TcP2β of individual phage selected from the fourth round of panning against this protein. Positive clones are shown (n=12). Cut-off (dashed line): 0·077. (C) Reactivity against peptide R13 of individual phage selected from the fourth round of panning against this peptide. Positive clones are shown (n=23). Cut-off (dashed line): 0·095.
Analysis of eluted phage displaying scFv antibodies
Individual clones (n=192) were selected from the fourth round of each panning for further analysis. Monoclonal phage were produced and analysed in Phage ELISA. As shown in Fig. 2B, 12 of the analysed phage were highly positive for TcP2β whereas 23 of the selected phage were positive for peptide R13 (Fig. 2C). The same antibodies were neither reactive against MBP nor TMVP peptide (data not shown).
To assess the quality and diversity of these positive clones, scFv-PCR and digestion with BstOI enzyme were performed. ScFv gene inserts were amplified with the ompseq and gback primers by colony-PCR. All the anti-TcP2β scFv had the expected fragment size (about 900 bp), while 17 out of 23 anti-R13 scFv had the full-length insert (Fig. 3A). The amplicons of the different recombinant antibodies were analysed by BstOI digestion and electrophoresis on 4% agarose gels (Fig. 3B). Eight anti-TcP2β scFv had distinct BstOI restriction fragment patterns. Amplicons derived from phage 17, 59, 81 and 163 had the same profile as well as amplicons from phage 6 and 42. Among the 17 anti-R13 scFv the same BstOI pattern was identified in 13 of them. Interestingly, the anti-TcP2β scFv 53 presented the same profile than these 13 anti-R13 scFv (Fig. 3B). Therefore, the BstOI fingerprint profiles established the diversity of the 8 scFv for TcP2β and 5 for R13 peptide meanwhile an scFv selected from both pannings was identified as the same one.

Fig. 3. scFv-PCR and BstOI fingerprint patterns of ELISA-positive clones obtained from panning against TcP2β and peptide R13. (A) ScFv genes were amplified from selected phage of round 4 by colony-PCR. The expected size for an scFv clone is about 900 bp. (B) Full-length insert clones were digested with BstOI enzyme. Lane M, 1 kb plus DNA marker (Invitrogen).
Expression and reactivity of the anti-TcP2β selected scFv
Different anti-P scFv were chosen for antibody expression. Antibody fragments without pIII were produced by introducing phagemid DNA into a non-suppressor strain of E. coli as TOP10. In 100 ml culture induction, 9 out of the 12 anti-TcP2β and anti-R13 selected different antibodies were expressed as soluble scFv (data not shown) but not all of them were reactive. Only 3 out of 9 expressed antibodies (anti-TcP2β scFv 42, 51 and 53) reacted with the corresponding antigen in ELISA and/or Western blot. It is worth noting that anti-TcP2β scFv 53 is the same as the 13 anti-R13 scFv mentioned before. As shown in Fig. 4A, Western blot of the 3 expressed reactive scFv showed a protein band with the expected size of approximately 30 kDa.

Fig. 4. Analysis of scFv expression and reactivity. (A) Expressed soluble scFv in E. coli (periplasmic extract) were analysed by Western blot. Indicated protein sizes were determined from broad range protein marker (M). (B) Detection of TcP2β by scFv 51 (periplasmic extract). As protein control, MBP was loaded in the same well. Positive control was the scFv C5 derived from the anti-TcP2β mAb 17·2 (Smulski et al. Reference Smulski, Labovsky, Levy, Hontebeyrie, Hoebeke and Levin2006). (C) Reactivity of the 3 expressed scFv against TcP2β and, peptides R13 and H13 by ELISA. scFv C10 and A4 are the recombinant anti-P antibodies derived from patients with SLE (Zampieri et al. Reference Zampieri, Mahler, Bluthner, Qiu, Malmegrim, Ghirardello, Doria, Van Venrooij and Raats2003). BSA and non-related TMVP peptide were used as controls. Positive control was the scFv C5 against TcP2β and peptide R13 (Smulski et al. Reference Smulski, Labovsky, Levy, Hontebeyrie, Hoebeke and Levin2006). * P<0·05. ** P<0·01.
Subsequently, scFv were used to probe Western blots of TcP2β (Fig. 4B). Notably, only scFv 51 recognized this recombinant. Thereafter we evaluated the reactivity of this scFv and the other antibodies by ELISA. As shown in Fig. 4C, scFv 42 and 53 that were not able to react with TcP2β in Western blots, did recognize the protein and its C-terminal end in ELISA. Moreover, they did not react with peptide H13 presenting the typical pattern of reactivity of anti-R13 antibodies in cChHD. On the contrary, scFv 51 reacted with the recombinant protein, but neither with R13 nor H13 (Fig. 4C). Recombinant anti-P antibodies derived from patients with SLE, namely scFv C10 and A4, reacted with both antigen TcP2β and peptide R13 as well as peptide H13. A monoclonal antibody against peptide R13 (scFv C5) that recognizes both, the recombinant protein and peptide R13 was used as a positive control for the experiment.
Sequence analysis
Comparison of the sequences with the closest known germline genes allowed us to carry out the identification of the V, D, and J gene segments used during somatic recombination to originate genes coding for the 3 reactive scFv. scFv 53 belongs to the VH4-4*07 family and both scFv 42 and 51 were encoded by the VH3-11*01 family. In contrast, autoanti-P antibodies from SLE patients (Zampieri et al. Reference Zampieri, Mahler, Bluthner, Qiu, Malmegrim, Ghirardello, Doria, Van Venrooij and Raats2003) belong to the VH3-23 and VH3-30 families. Comparison of the amino acid sequences of the cloned immunoglobulins showed no sequence similarities (Fig. 5A and B).
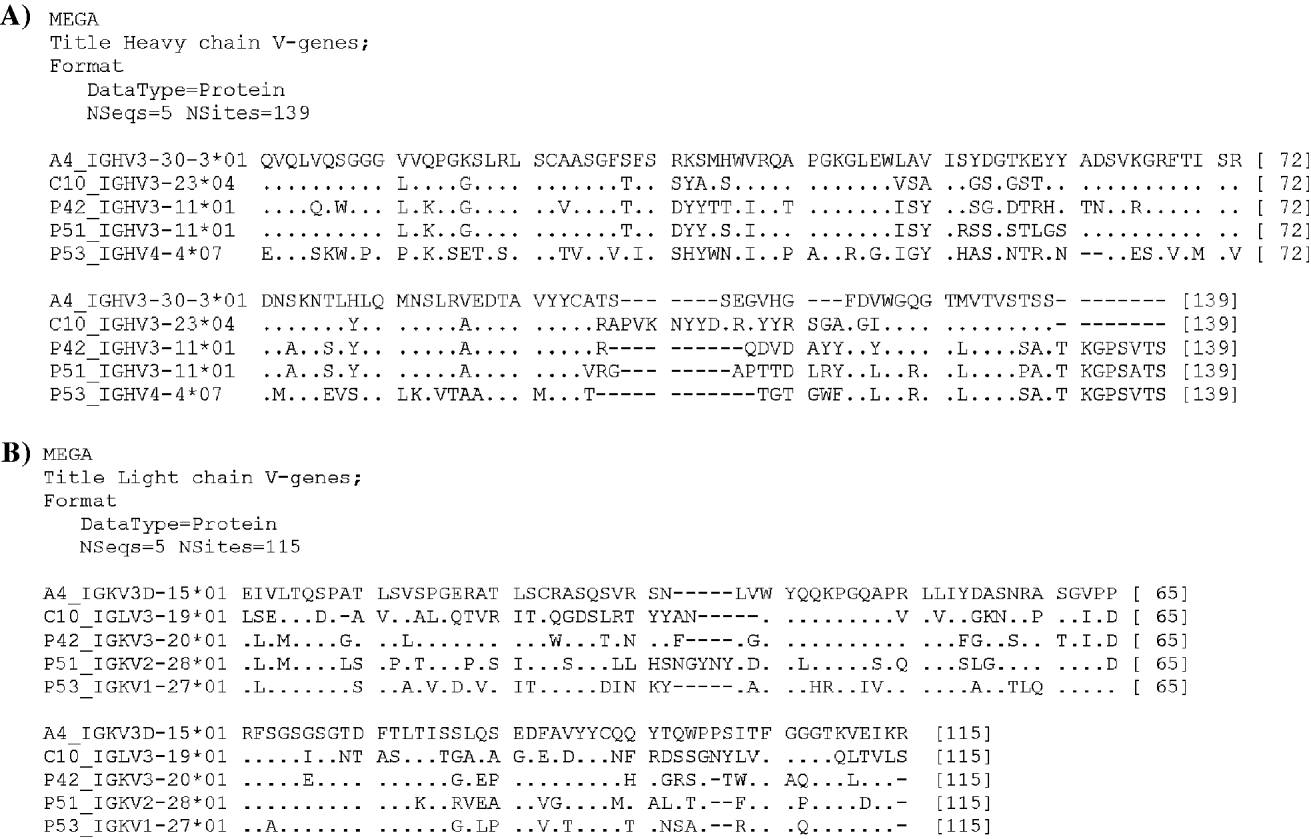
Fig. 5. Amino acid sequence alignment of anti-TcP2β scFv 42, 51 and 53 (cChHD library) and anti-human ribosomal P protein scFv C10 and A4 (SLE library) (Zampieri et al. Reference Zampieri, Mahler, Bluthner, Qiu, Malmegrim, Ghirardello, Doria, Van Venrooij and Raats2003). VH (A) and VL (B) regions are shown. Alignment analyses were conducted using MEGA version 4 (Tamura et al. 2007). Amino acids that are identical are symbolized by dots, and the absence of amino acid by dashes.
The main features of these antibodies are summarized in Table 2. CDRH3 length for the 3 anti-TcP2β scFv was 39 bp for scFv 42, 42 bp for scFv 51 and 33 bp for scFv 53. The SLE anti-P antibodies had CDRH3 lengths of 33 bp and 66 bp for A4 and C10, respectively.
Table 2. Summarized features of anti-TcP2β scFv 42, 53 and 51 selected from cChHD library and anti-human ribosomal P protein scFv C10 and A4 selected from SLE libraries (Zampieri et al. Reference Zampieri, Mahler, Bluthner, Qiu, Malmegrim, Ghirardello, Doria, Van Venrooij and Raats2003)

Using a 98% germline homology as cut off (Kienle et al. Reference Kienle, Krober, Katzenberger, Ott, Leupolt, Barth, Moller, Benner, Habermann, Muller-Hermelink, Bentz, Lichter, Dohner and Stilgenbauer2003; Arons et al. Reference Arons, Sunshine, Suntum and Kreitman2006), the 3 cChHD antibodies and 1 of the anti-P antibodies, A4, were considered to be mutated. In the case of the 3 anti-TcP2β scFv, the germline homology was 86·67% for scFv 53, 92·71% for scFv 42 and 96·88% for scFv 51. The anti-P scFv A4 from SLE patients presented 93·75% germline homology.
Using the multinomial distribution model equation (Lossos et al. Reference Lossos, Tibshirani, Narasimhan and Levy2000), only scFv 42 and 53 may be considered to be hypermutated showing clear evidence of antigen-driven selection (Table 2).
DISCUSSION
We have constructed a phage-displayed scFv antibody library (2×109 cfu/μg DNA) derived from the bone marrow of a cChHD patient. This library is formed by around 1·85×109 phage (92·5% functional clones of the original library). This magnitude order allowed us to identify anti-TcP2β and anti-R13 scFv.
Three recombinant human antibodies were obtained, scFv 42, 51 and 53 that reacted with TcP2β. Two of them represented the reactive profile that defined an anti-R13 antibody, with a strong anti-TcP2β reactivity and no reactivity to the human H13 peptide, the human version of the C-terminal end of the human ribosomal P proteins. This peptide contains only 1 amino acid change compared to the T. cruzi R13 sequence (H13: EESDDDMGFGLFD) (Elkon et al. Reference Elkon, Bonfa, Llovet, Danho, Weissbach and Brot1988). This typical pattern of reactivity of anti-R13 antibodies has been observed in cChHD, in experimental infections and in some animals immunized with the TcP2β (Kaplan et al. Reference Kaplan, Ferrari, Lopez-Bergami, Mahler, Levitus, Chiale, Hoebeke, Van Regenmortel and Levin1997; Lopez Bergami et al. Reference Lopez Bergami, Scaglione and Levin2001, Reference Lopez Bergami, Gomez, Levy, Grippo, Baldi and Levin2005; Mahler et al. Reference Mahler, Hoebeke and Levin2004; Labovsky et al. Reference Labovsky, Smulski, Gómez, Levy and Levin2007). The strong R13 reactivity and no H13 reactive pattern were clearly different from the reactivity of the anti-P autoantibodies that recognized both peptides equally. In addition to this, the anti-TcP2β response was also characterized by the existence of a second epitope that is not contained within R13, but enclosed within the complete TcP2β protein (scFv 51). The existence of such an epitope, probably associated with a specific conformation of the TcP2β, had been suggested by Levin et al. (Reference Levin, Rossi, Levitus, Mesri, Bonnefoy, Kerner and Hontebeyrie-Joskowicz1990). Thus, our findings confirmed the very specific nature of the antibody response against this parasite protein.
Years ago, characterization of the anti-P response in cChHD patients led to identification of antibodies with putative auto-reactive properties that were compared to anti-P autoantibodies considered to be markers of SLE (Mesri et al. Reference Mesri, Levitus, Hontebeyrie-Joskowicz, Dighiero, Van Regenmortel and Levin1990). Moreover, several authors suggested a possible mechanistic link between Chagas disease and SLE and other autoimmune disorders (Mesri et al. Reference Mesri, Levitus, Hontebeyrie-Joskowicz, Dighiero, Van Regenmortel and Levin1990; Levitus et al. Reference Levitus, Hontebeyrie-Joskowicz, van Regenmortel and Levin1991; Skeiky et al. Reference Skeiky, Benson, Parsons, Elkon and Reed1992). For this reason, sequence alignment analysis of V genes from selected anti-TcP2β scFv and anti-P autoantibodies from SLE patients was also performed and showed no similarities among these antibodies (Lefranc, Reference Lefranc2003). The 3 analysed anti-TcP2β antibodies belong to the most representative VH families in cChHD repertoire (VH3 and VH4) (Grippo et al. Reference Grippo, Mahler, Elias, Cauerhff, Gómez, Tentori, Ruiz, Vigliano, Laguens, Berek and Levin2009). Of note is the finding that no autoimmune-related genes such as VH4-34 or VH3-23 were observed among these anti-P scFv from the cChHD patient. Notably, the anti-P autoantibodies from SLE patients (Zampieri et al. Reference Zampieri, Mahler, Bluthner, Qiu, Malmegrim, Ghirardello, Doria, Van Venrooij and Raats2003) belong to the VH3-23 and VH3-30 families found to encode autoantibodies (Brezinschek et al. Reference Brezinschek, Brezinschek and Lipsky1995; Voswinkel et al. Reference Voswinkel, Weisgerber, Pfreundschuh and Gause2001). Moreover, autoanti-P scFv C10 derived from SLE patients was not mutated and belongs to one of the reference VH genes used for autoantibodies, VH3-23 (Voswinkel et al. Reference Voswinkel, Weisgerber, Pfreundschuh and Gause2001; Garcia-Muñoz et al. Reference García-Muñoz, Panizo, Bendandi and Llorente2009).
The CDRH3 length of these antibodies is shorter than those reported for other infectious diseases as has previously been described for Chagas disease (Grippo et al. Reference Grippo, Mahler, Elias, Cauerhff, Gómez, Tentori, Ruiz, Vigliano, Laguens, Berek and Levin2009). It has been suggested that specific antibodies for small molecules (haptens) have concave antigen-binding sites, involving larger CDRs, while those that bind larger molecules such as proteins, tend to have flattened antigen-binding sites, e.g. shorter CDRs (Vargas-Madrazo et al. Reference Vargas-Madrazo, Lara-Ochoa and Almagro1995). It might be proposed that in the anti-TcP2β response, one would find a skew of VH regions towards shorter CDR3 loops when compared to the response against the whole parasite. In fact, the reactive scFv 42, 51 and 53 presented CDR3 lengths that were clearly below the median calculated for the anti-T. cruzi VH genes (Grippo et al. Reference Grippo, Mahler, Elias, Cauerhff, Gómez, Tentori, Ruiz, Vigliano, Laguens, Berek and Levin2009). Interestingly, mutational analysis showed that scFv 42 and 53 were the only hypermutated human mAbs selected from the cChHD library and the only two that recognized the peptide R13. Moreover, scFv 53 was the one selected from both pannings and the most representative among anti-R13 antibodies. What is more, this scFv presented the highest reactivity in ELISA and the main difference in homology with the corresponding VH germline gene.
In this study, a combinatorial phage display scFv library was constructed starting from bone marrow of a patient with cChHD. Phage bearing specific scFv fragments for the parasite antigen were selected successfully by panning, which allowed us to obtain, for the first time, human mAbs against T. cruzi ribosomal P2β protein. The characterization of these anti-TcP2β mAbs confirmed the typical pattern of reactivity of the anti-R13 antibodies observed in cChHD patients, as well as the existence of a second epitope not contained in the C-terminal end but enclosed in the TcP2β protein. In addition to this, the reactive profile and V gene usage of these human anti-TcP2β mAbs were clearly different from the anti-P autoantibodies derived from SLE patients. Furthermore, this Chagas patient-derived phage antibody library is a powerful tool to isolate specific immunological components of human origin against many different parasite antigens. The available spectrum of mAbs against T. cruzi will be useful in both the characterization of the parasite's antigenic composition and the human hosts' immune response to T. cruzi infection. In conclusion, antibody phage display technology is a potential approach for the study of antigen-antibody interactions used to further elucidate the pathogenesis of Chagas disease.
ACKNOWLEDGMENTS
This paper is dedicated to the memory of Dr Mariano J. Levin (1951–2010).
The authors are grateful to the CSHL phage display course team, Dr Carlos Barbas 3rd (Department of Chemistry, The Scripps Research Institute, La Jolla, CA, USA), Dr Donald Siegel (University of Pennsylvania Medical Center, PA, USA) and Dr Gregg Silverman (Laboratory of B-cell Immunobiology, University of California, San Diego, La Jolla, CA, USA) for providing us with both the knowledge for the construction of the phage display library as well as for the pComb3X vector system for cloning the human library. We are also thankful to Dr Jos Raats (ModiQuest B.V. and Department of Biomolecular Chemistry, Nijmegen Centre for Molecular Life Sciences, Radboud University Nijmegen, The Netherlands) for kindly providing the vectors for the expression of anti-P autoantibodies A4 and C10 derived from SLE patients. We thank Sonia Lafon for excellent technical assistance.
FINANCIAL SUPPORT
This work was supported mainly by an International Research Grant from the Howard Hughes Medical Institute (Chevy Chase, MD, USA). This research was also supported by grants from the World Health Organization/Special Program for Research and Training in Tropical Diseases; the Argentinean National Council for Science and Technology (CONICET), the University of Buenos Aires, Health and Social Action Office-fellowship Ramón Carrillo-Arturo Oñativia, the National Agency of Scientific and Technological Promotion (FONCYT BID 1201/OC-AR, PICT 01–14389 and BID 1728/ OC-AR PICT 2006 No. 2439) and partly supported by the Bunge & Born Foundation of Argentina.







