


INTRODUCTION
Leishmaniases are a group of diseases caused by flagellate parasites of the genus Leishmania Ross, 1903 (Kinetoplastida: Trypanosomatidae). They are endemic in approximately 100 countries in Africa, America, Asia and Europe, and cause more than one million cases and several thousand deaths each year (Alvar et al. Reference Alvar, Vélez, Bern, Herrero, Desjeux, Cano, Jannin and Boer2012). More than 50 Leishmania species have been described to date, among which approximately 20 are known to infect humans (Maroli et al. Reference Maroli, Feliciangeli, Bichaud, Charrel and Gradoni2013; Akhoundi et al. Reference Akhoundi, Kuhls, Cannet, Votýpka, Marty, Delaunay and Sereno2016). These may cause various clinical forms (e.g. localized cutaneous, disseminated cutaneous, mucocutaneous or visceral leishmaniasis) and respond differently to treatment (Copeland and Aronson, Reference Copeland and Aronson2015). Furthermore, each Leishmania species is characterized by a specific transmission cycle involving distinct sand fly vectors and reservoir hosts (Lainson and Shaw, Reference Lainson and Shaw2010; Maroli et al. Reference Maroli, Feliciangeli, Bichaud, Charrel and Gradoni2013; Ready, Reference Ready2013). Therefore, in areas where several Leishmania species are found in sympatry or when imported cases can occur, reliable identification of strains is crucial for medical management and eco-epidemiological studies.
For more than 40 years, multilocus enzyme electrophoresis (MLEE) has allowed considerable advances in the delineation and classification of Leishmania species (Gardener and Howells, Reference Gardener and Howells1972; Kreutzer et al. Reference Kreutzer, Semko, Hendricks and Wright1983; Rioux et al. Reference Rioux, Lanotte, Serres, Pratlong, Bastien and Perieres1990; Thomaz-Soccol et al. Reference Thomaz-Soccol, Lanotte, Rioux, Pratlong, Martini-Dumas and Serres1993). Nevertheless, this technique is time-consuming and relies on constraining steps of parasite isolation and culture. Polymerase chain reaction (PCR)-based methods are increasingly used as alternatives to MLEE for Leishmania typing because they are cost and time-effective and can be performed on small quantities of material. In particular, restriction fragment length polymorphism (RFLP) analyses (Marfurt et al. Reference Marfurt, Nasereddin, Niederwieser, Jaffe, Beck and Felger2003; Rotureau, Reference Rotureau2006; Koarashi et al. Reference Koarashi, Cáceres, Saca, Flores, Trujillo, Alvares, Yoshimatsu, Arikawa, Katakura, Hashiguchi and Kato2016) and direct comparisons of DNA sequences (Zelazny et al. Reference Zelazny, Fedorko, Li, Neva and Fischer2005; Marco et al. Reference Marco, Uezato, Mimori, Barroso, Korenaga, Nonaka, Basombrío, Taranto and Hashiguchi2006; de Almeida et al. Reference de Almeida, Steurer, Koru, Herwaldt, Pieniazek and da Silva2011) have been widely employed in recent years. Various genomic regions have been targeted for molecular identification of Leishmania, in a quest for the best compromise between sensitivity, specificity and taxonomic coverage (Akhoundi et al. Reference Akhoundi, Downing, Votýpka, Kuhls, Lukeš, Cannet, Ravel, Marty, Delaunay, Kasbari, Granouillac, Gradoni and Sereno2017), but no real consensus has been reached in this regard.
Leishmania parasites, such as all kinetoplastids, are characterized by a unique mitochondrion containing a remarkably large and dense genome: the kinetoplast. Kinetoplast DNA (kDNA) represents 10–20% of total cellular DNA (Simpson, Reference Simpson1987) and is composed of two types of circular molecules interlinked in a concatenated network: maxicircles and minicircles. Maxicircles are c. 20 kb long, present in a few tens of copies, and are analogous to the mitochondrial genome of other eukaryotes. Minicircles are c. 800 bp long, present in several thousands of copies. They encode for guide RNAs (gRNAs) involved in the maturation of maxicircle's messenger RNAs through an RNA editing mechanism (Read et al. Reference Read, Lukeš and Hashimi2015). Due to their extremely high copy number, minicircles are ideal targets for highly sensitive detection of Leishmania. Furthermore, the presence of well-conserved sequence blocks (CSBs) in their replication origin (Jensen and Englund, Reference Jensen and Englund2012) allows the design of PCR primers with wide taxonomic coverage (Noyes et al. Reference Noyes, Reyburn, Bailey and Smith1998; Ceccarelli et al. Reference Ceccarelli, Galluzzi, Migliazzo and Magnani2014).
On the other hand, the analysis of minicircle sequences for Leishmania species identification is difficult. The pool of minicircles (minicirculome) present in each parasitic cell encompasses a set of distinct minicircle versions, referred to as classes, encoding different gRNAs (Hajduk and Ochsenreiter, Reference Hajduk and Ochsenreiter2010). Except in the CSBs, high sequence variability is found among minicircle classes (Brewster and Barker, Reference Brewster and Barker2002), resulting in difficulties for their analysis (de Oliveira Ramos Pereira and Brandão, Reference de Oliveira Ramos Pereira and Brandão2013). Furthermore, there are redundant gRNAs that encode the same editing information, and the number and identity of minicircle classes vary greatly between species and even between strains of the same species (Simpson, Reference Simpson1997; Gao et al. Reference Gao, Kapushoc, Simpson, Thiemann and Simpson2001). These features make classical DNA-based identification methods difficult to perform. Thus, in many studies, minicircle-based PCR assays are employed as highly sensitive screening tests for Leishmania detection, while other genomic regions are targeted in a second step for the identification of positive samples (Cássia-Pires et al. Reference Cássia-Pires, Boité, D'Andrea, Herrera, Cupolillo, Jansen and Roque2014; Richini-Pereira et al. Reference Richini-Pereira, Marson, Hayasaka, Victoria, da Silva and Langoni2014; Berzunza-Cruz et al. Reference Berzunza-Cruz, Rodríguez-Moreno, Gutiérrez-Granados, González-Salazar, Stephens, Hidalgo-Mihart, Marina, Rebollar-Téllez, Bailón-Martínez, Balcells, Ibarra-Cerdeña, Sánchez-Cordero and Becker2015; Pereira Júnior et al. Reference Pereira Júnior, Teles, de Azevedo dos Santos, de Souza Rodrigues, Marialva, Pessoa and Medeiros2015).
The characterization of Leishmania minicirculomes used to require isolation and cloning steps prior to sequencing (Lee et al. Reference Lee, Tarn and Chang1993; Brewster and Barker, Reference Brewster and Barker2002; Telleria et al. Reference Telleria, Lafay, Virreira, Barnabé, Tibayrenc and Svoboda2006; Rodrigues et al. Reference Rodrigues, da Silva Soares, Werkhäuser, de Brito, Fernandes, Abath and Brandão2013). Here, we show that high-throughput sequencing of minicircle PCR amplicons may be an efficient alternative in this regard. We then present a barcoding-like approach based on minicircle sequence comparisons, which aims at combining sensitive detection and reliable identification of Leishmania spp. The method is evaluated for a use in the Neotropical region, where a high diversity of Leishmania species may be found in sympatry (Lainson and Shaw, Reference Lainson and Shaw2010).
MATERIALS AND METHODS
Leishmania strains and DNA extraction
Eighteen Leishmania strains were selected, representative of both Leishmania subgenera and of all major species complexes found in the neotropics (Table 1). They belonged to six species (as identified by MLEE or PCR–RFLP; (Simon et al. Reference Simon, Veron and Carme2010)): L. (Viannia) braziliensis, L. (V.) guyanensis, L. (V.) lainsoni, L. (V.) naiffi, L. (Leishmania) amazonensis and L. (L.) infantum. When possible, we included several strains isolated in distant locations for each Leishmania species. Leishmania parasites were cultured as previously described (Faye et al. Reference Faye, Bañuls, Bucheton, Dione, Bassanganam, Hide, Dereure, Choisy, Ndiaye, Konaté, Claire, Senghor, Faye, Sy, Niang, Molez, Victoir, Marty, Delaunay, Knecht, Mellul, Diedhiou and Gaye2010). DNA was extracted from culture pellets using the Pure Link Genomic DNA Kit (Life Technologies, Saint-Aubin, France).
Table 1. List of the strains used in this study
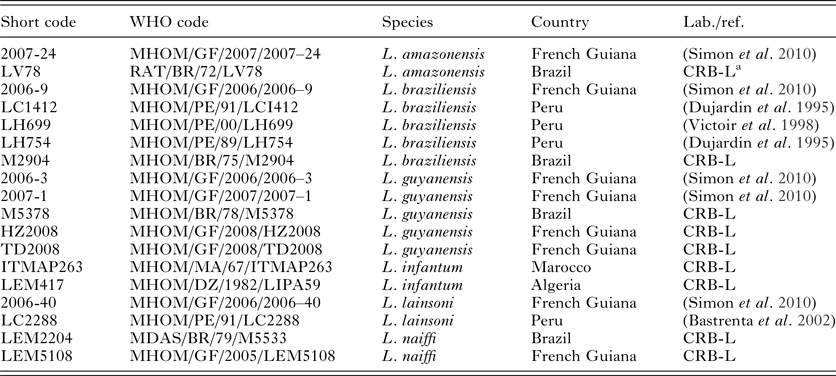
FG, French Guiana; B, Brazil; P, Peru; M, Marocco; A, Algeria.
a Centre de Ressources Biologiques des Leishmania, Monptellier, France.
PCR amplification
PCR amplification targeted the c. 120 pb portion of kDNA minicircle known as the minicircle conserved region. We used PCR primers located within CSB1 and CSB3, similar to previously published ones (Fig. 1; Roque et al. Reference Roque, Cupolillo, Marchevsky and Jansen2010; Weirather et al. Reference Weirather, Jeronimo, Gautam, Sundar, Kang, Kurtz, Haque, Schriefer, Talhari, Carvalho, Donelson and Wilson2011), but further adjusted based on a large set of complete minicircle sequences (leishmini-F: 5′-GGKAGGGGCGTTCTGC-3′; leishmini-R: 5′-STATWTTACACCAACCCC-3′). Our aim was to ensure that all minicircle classes will be amplified in every Leishmania spp.
Amplification was performed in 20 µL mixtures containing 2 µL of DNA template, 10 µL of AmpliTaq Gold PCR Master Mix® (5 U.μL−1; Applied Biosystems, Foster City, CA, USA), 2·5 µL of each primer (5 µ m), and nuclease-free water (Promega, Madison, WI, USA). The PCR mixture was denatured at 95 °C (10 min) and followed by 35 cycles of 30 s at 95 °C, 30 s at 60 °C and 15 s at 72 °C, completed at 72 °C for 5 min. To enable the sequencing of multiple PCR products in a single high-throughput sequencing run, tags of eight base pairs with at least five differences between them were added at the 5′ end of each primer (Binladen et al. Reference Binladen, Gilbert, Bollback, Panitz, Bendixen, Nielsen and Willerslev2007). In order to evaluate the frequency of tag-switching events, which may result in some sequences being assigned to the wrong sample (Schnell et al. Reference Schnell, Bohmann and Gilbert2015), we only used one out of two available tag combinations, as suggested by (Esling et al. Reference Esling, Lejzerowicz and Pawlowski2015). PCR sensitivity was checked on a serial dilution of Leishmania DNA followed by electrophoresis and visualization of PCR product in a 2% agarose gel containing SYBR Safe stain (Invitrogen) under UV light.
High-throughput sequencing of minicircle amplicons
PCR products obtained from the 18 Leishmania strains were pooled and sent to the GeT-PlaGe core facilities of GenoToul (Toulouse, France) for library construction and sequencing on an Illumina Hiseq 3000 platform (Illumina, San Diego, CA, USA). Quality filtering was performed with the Consensus Assessment of Sequence and Variation (CASAVA) pipeline. Sequence data were stored on the NG6 platform (Mariette et al. Reference Mariette, Escudié, Allias, Salin, Noirot, Thomas and Klopp2012).
Sequencing reads were analysed with OBITOOLS (Boyer et al. Reference Boyer, Mercier, Bonin, Le Bras, Taberlet and Coissac2016), as described elsewhere (Kocher et al. Reference Kocher, Thoisy, Catzeflis, Huguin, Valiere, Zinger, Bañuls and Murienne2017a, Reference Kocher, Gantier, Gaborit, Zinger, Holota, Valiere, Dusfour, Girod, Bañuls and Murienne2017b). Paired-end reads were aligned and merged, taking into account the Phred quality scores for consensus construction. Merged reads were assigned to a given sample based on the primer tags with two mismatches allowed. Low-quality reads (exhibiting alignment scores <50, containing Ns or shorter than 50 bp) were discarded and then dereplicated in each sample. To limit errors arising from tag-switching events, we discarded all sequences that had coverage below that of the most abundant sequence found with an unexpected tag combination. The bash script used for these bioinformatic steps is available in the Supplementary Material.
Minicircle sequence analysis and use for species identification
Because multiple alignments of minicircle sequences are problematic (de Oliveira Ramos Pereira and Brandão, Reference de Oliveira Ramos Pereira and Brandão2013), we performed pairwise alignments of each sequence pair based on longest common subsequences (Needleman–Wunsch alignment with a score of one for matches and zero for mismatches and gaps). Raw distances were then used to generate a neighbour-joining (NJ) tree with the R package ‘ape’ (Saitou and Nei, Reference Saitou and Nei1987; Paradis et al. Reference Paradis, Claude and Strimmer2004; R Core team, 2014), to visualize clustering patterns among Leishmania strains and species. Distance statistics were computed using the R package ‘spider’ (Brown et al. Reference Brown, Collins, Boyer, Lefort, Malumbres-Olarte, Vink and Cruickshank2012). In order to provide an alternative visualization of the data, we constructed a haplotype network. Minicircle sequences were aligned using muscle v. 3.8.31 (Edgar, Reference Edgar2004) and highly variable regions were removed using Gblocks 0·91b (Castresana, Reference Castresana2000) with default parameters. The network was constructed using PopART (popart.otago.ac.nz) with the minimum spanning network method (Bandelt et al. Reference Bandelt, Forster and Röhl1999).
Our aim was then to use a barcoding-like strategy to perform Leishmania species identifications based on minicircle sequences. For each Leishmania strain, several minicircle classes were expected to be amplified and recovered by high-throughput sequencing. Therefore, taxonomic assignments of Leishmania spp. required the use of a multilocus approach. However, contrary to multilocus barcoding, where several markers are sequenced separately (Fazekas et al. Reference Fazekas, Burgess, Kesanakurti, Graham, Newmaster, Husband, Percy, Hajibabaei and Barrett2008; Dufour et al. Reference Dufour, Pons, Collinson, Gernigon, Dies, Sourrouille and Crochet2017), we did not have a priori knowledge of the identity of each minicircle fragments retrieved. It was thus not possible to perform sequence alignment for each minicircle classes independently. Additionally, the set of minicircle classes may vary between strains, even within the same Leishmania species (Simpson, Reference Simpson1997; Gao et al. Reference Gao, Kapushoc, Simpson, Thiemann and Simpson2001). Thus, there was no guaranty that each minicircle sequence recovered from a strain would find a homologue among those of other conspecific references. Furthermore, some minicircle classes may be species-specific, while others are shared between species (Brewster and Barker, Reference Brewster and Barker2002). Therefore, we had to consider that only a fraction of sequences would be informative for taxonomic assignments.
As a consequence, when comparing a set of minicircle sequences from an unidentified Leishmania strain to that of reference strains, each may either (i) match closely with one or several homologous references all belonging to the same species (i.e. specific match); (ii) match closely with several homologous references from various species (i.e. unspecific match); or (iii) not match closely with any available reference (i.e. no homologue, Fig. 2). Eventually, the taxonomic assignment of the strain should be based on the subset of minicircles for which specific matches were found.

Fig. 1. Structural organization of Leishmania kinetoplast minicircles. The three conserved sequence blocks (CSBs) are indicated in white boxes. Blue arrows indicate the binding sites of PCR primers used to amplify the c. 120-bp-long fragment within the conserved region.
In practice, for a given unidentified strain, we used the ecotag program (included in the OBITOOLS package) to perform the taxonomic assignment of each minicircle sequence. ecotag includes a ‘last common ancestor algorithm’ that allows one to distinguish between specific and unspecific matches. The strain was then taxonomically assigned according to the most frequent specific match, after weighting by sequence abundances (Fig. 2). The proportion of specific matches supporting the resulting identification was used as an identification score.

Fig. 2. Schematization of the pipeline used for the identification of Leishmania spp. based on high-throughput sequencing of c. 80-bp-long kDNA minicircle amplicons.
To evaluate the accuracy of the method, we performed a ‘leave one out’ testing. Each strain (i.e. the full set of minicircle sequences) was consecutively considered unknown and identified using the method described above with the remaining strains as references. Bash and R scripts used to perform the taxonomic assignment of strains and to compute identification scores are available in the Supplementary Material.
RESULTS
High-throughput sequencing of minicircle amplicons
PCR amplification of kDNA minicircles allowed the detection of all Leishmania species down to 1 fg of template DNA (i.e. below 10−2 parasite equivalent; see Supplementary material). High-throughput sequencing of PCR products allowed obtaining a total of 10 347 227 pairs of 150 bp long reads. After paired-end merging, quality filtering removed 0·23% of reads. The 670 890 unique sequences remained after reading dereplication. The most abundant sequence assigned to a non-used tag combination had a coverage of 1172, which was used as a threshold to remove sequences potentially originating from tag-switching events. Remaining sequences had an average read coverage of 4380. Eventually, the number of minicircle sequences retrieved ranged from 10 to 84 depending on the strain, for an average of 37·2 (Table 3; GenBank acc. KY699529–KY700198). Minicircle amplicon length ranged from 80 to 82 bp.
Minicircle sequence analysis and use for species identification
Based on the pairwise alignment of minicircle sequences, average intraspecific and interspecific raw genetic distances were 11·5 and 26·4%, respectively. The highest mean intraspecific distance was found in L. amazonensis (18·9%; Table 2), while the lowest was found in L. naiffi, L. guyanensis and L. braziliensis (10·3, 10·4 and 10·6%, respectively). In the NJ tree, minicircle sequences of each strain did not form single monophyletic clusters, but were rather distributed in several clusters comprising sequences of other strains, generally belonging to the same species (Fig. 3). This pattern was also highlighted by very similar mean intrastrain and interstrain genetic distance within each species (Table 2). At the species level, several patterns were observed. Almost all sequences were grouped in single species-specific clusters for L. amazonensis, L. infantum and L. lainsoni. For the three other species (L. naiffi, L. braziliensis and L. guyanensis), minicircle sequences formed several interleaved clusters. Some of these were well delimited and species-specific, while others included a mix of species (for L. braziliensis and L. guyanensis especially). After multiple alignments of minicircle sequences and the removal of highly variable sequence blocks, 37 nucleotide positions were selected to construct the minimum spanning network (Fig. 4). This allows an alternative visualization of the previously described pattern. L. amazonensis and L. infantum did not share any minicircle haplotype with other species (except for one in the case of L. infantum) and formed separate clusters. On the contrary, in the Viannia subgenus, many minicercles haplotypes were shared between species, especially between L. guyanensis and L. braziliensis. L. lainsoni appeared to share most of its minicircle haplotypes with other species of the Viannia subgenus, while the majority of L. naiffi haplotypes were monospecific.

Fig. 3. Neighbour-joining tree based on pairwise alignments of c. 80-bp-long kDNA minicircle fragments. Only the 20 most abundant sequences are included for each strain to allow visualization. The colours refer to Leishmania species. Each sequence is referenced with its strain ID.

Fig. 4. Minimum spanning network of minicircle sequences after removal of highly variable regions.
Table 2. Minicircle sequence distance statistics by Leishmania species

The leave-one-out procedure showed that, using the method presented here, all strains would have been correctly identified if considered unknown (Table 3). On average, for each strain, 84·8% of minicircle sequences matched on species-specific references, and these were indicative of the correct Leishmania species in 97·85% of cases. Most strains (10/18) were identified with an identification score of 100%, including all L. amazonensis, L. infantum, L. lainsoni and L. naiffi strains. Some minicircle sequences of L. braziliensis were identified as L. guyanensis and vice versa, which resulted in identification scores below 100%. However, most L. braziliensis and L. guyanensis were identified with a score above 95%. The most ambiguous identifications were that of strains M5378 and HZ2008 (scores = 90·4 and 88·4%, respectively), but these were still correctly identified as L. guyanensis.
Table 3. Taxonomic identification of the strain using a leave-one-out procedure

LA, L. amazonensis; LB, L. braziliensis; LG, L. guyanensis; LI, L. infantum; LL, L. lainsoni; LN, L. naiffi; NS, not specifically assigned.
a Proportion of sequences reads assigned to each species.
DISCUSSION
The kDNA minicircle conserved region is known to be an ideal target for the detection of all Leishmania spp. with high sensitivity (Akhoundi et al. Reference Akhoundi, Downing, Votýpka, Kuhls, Lukeš, Cannet, Ravel, Marty, Delaunay, Kasbari, Granouillac, Gradoni and Sereno2017). This was further confirmed by our results, since PCR amplification was successful for the six Leishmania species belonging to both Viannia and Leishmania subgenera, down to an extremely low DNA concentration. On the other hand, we would like to stress that other non-Leishmania kinetoplastids could probably be detected with the same PCR assay due to the high conservation of CSBs, which should not be used for the specific diagnosis without confirmation by sequencing.
Despite the usefulness of kDNA minicircles for Leishmania detection, classical DNA-based identification methods may hardly be applicable for these peculiar genomic molecules. One of the reasons is that several distinct minicircle classes coexist in each parasitic cell. Even if all minicircles can be amplified simultaneously with the same PCR primers located in the CSBs, the comprehensive characterization of minicircle population used to require isolation and cloning of PCR products prior to sequencing (Lee et al. Reference Lee, Tarn and Chang1993; Brewster and Barker, Reference Brewster and Barker2002; Telleria et al. Reference Telleria, Lafay, Virreira, Barnabé, Tibayrenc and Svoboda2006; Rodrigues et al. Reference Rodrigues, da Silva Soares, Werkhäuser, de Brito, Fernandes, Abath and Brandão2013). High-throughput sequencing allows bypassing these laborious tasks and will likely contribute to fulfil important methodological gaps in kinetoplastid genomics. By sequencing single PCR products, we were able to easily retrieve significant numbers of distinct minicircle amplicons from each strain, reflecting the diversity of minicircle classes.
Another difficulty comes with the variability in the number and identity of minicircle classes found among strains, even within the same species. Therefore, we used a barcoding-like strategy that does not rely on a priori knowledge of minicircle homology. Our results show that, using our method, every strain would have been correctly identified, even when conspecific reference strains were isolated from very distant geographic locations. This highlights the robustness of the approach for the identification of all major species complexes found in the New World. Other genetic markers can provide such level of taxonomic resolution (Akhoundi et al. Reference Akhoundi, Downing, Votýpka, Kuhls, Lukeš, Cannet, Ravel, Marty, Delaunay, Kasbari, Granouillac, Gradoni and Sereno2017), but thanks to the very high copy number of kDNA minicercles, the present method may be employed in a single assay for sensitive Leishmania detection and Leishmania spp. identification. The inclusion of additional closely related Leishmania species would be necessary to assess whether the method is reliable beyond the species complex level. Further effort is also needed to extend the present work to a wider range of parasites, including Old World Leishmania spp. as well as other Trypanosomatidae. In the future, the development of similar strategies targeting the minicircle variable region may allow the typing of strains at a finer resolution. Indeed, kDNA minicircle fingerprinting has long been known as a valuable tool for describing and tracking Leishmania genetic diversity, with a discriminatory power superior to that of MLEE (Angelici et al. Reference Angelici, Gramiccia and Gradoni1989; Botilde et al. Reference Botilde, Laurent, Quispe Tintaya, Chicharro, Cañavate, Cruz, Kuhls, Schönian and Dujardin2006).
One limitation of the approach is that it may be unable to identify Leishmania hybrids (Bañuls et al. Reference Bañuls, Guerrini, Pont, Barrera, Espinel, Guderian, Echeverria and Tibayrenc1997; Ravel et al. Reference Ravel, Cortes, Pratlong, Morio, Dedet and Campino2006), or to detect the presence of several Leishmania spp. in the same sample [e.g. in the case of a coinfection, or when analysing pools of sand flies (Kocher et al. Reference Kocher, Gantier, Gaborit, Zinger, Holota, Valiere, Dusfour, Girod, Bañuls and Murienne2017b)]. This is because, even in one single Leishmania strain, minicircle sequences may give conflicting taxonomic signals (i.e. some sequences being identified as belonging to a given species and other being identified as belonging to another). We showed that considering only the majority taxonomic signal allowed robust species identifications. However, mixed infections or hybrid strains could hardly be distinguished from the intrastrain mixed signal. The constitution of more comprehensive and local minicircle reference databases will probably narrow the spectrum of species-specific reference minicircle sequences, which could help resolve this issue.
Another drawback of the method is that high-throughput sequencing remains relatively costly unless large batches of samples are analysed, and that several days or weeks may be required for laboratory work to bioinformatic treatment of the data. Such an approach is therefore not adapted for all applications yet. In particular, the present assay is clearly not adapted for routine diagnostic. However, it can already be useful for one-time studies involving the analysis of large number of samples. Furthermore, given the rapid evolution of high-throughput sequencing technologies and their increasing accessibility, this type of approach could be used more widely in a near future.
Our analyses revealed patterns of sequence clustering that are remarkably congruent with the current hypothesis of Leishmania relationships and timings of speciation (Harkins et al. Reference Harkins, Schwartz, Cartwright and Stone2016). In particular, the observation of the NJ tree and minimum spanning network seems to indicate that the presence of shared or homologue minicircle classes between species tend to decrease when the given species are more distantly related. More extensive studies based on similar high-throughput sequencing approaches may provide precious insights into the evolutionary aspects of kinetoplast minicircle networks. Additionally, the method presented here could be adapted to acquire knowledge in fundamental biological processes of these parasites. Minicircle analyses in Trypanosoma brucei and T. cruzi allowed to evidence clear signatures of intraspecific recombination (Gibson et al. Reference Gibson, Crow and Kearns1997). Population structure in Leishmania still needs to be further detailed, especially as to the occurrence of genetic exchanges (Rougeron et al. Reference Rougeron, De Meeûs and Bañuls2017). High-throughput sequencing of minicircle amplicons may represent a powerful tool in this regard.
Concluding remarks
In this study, we showed the efficiency of high-throughput sequencing to recover various minicircle classes simultaneously amplified by PCR. Despite the known difficulties for minicircle sequence analysis, we developed a barcoding-like strategy that proved to be robust for all major Leishmania species complexes found in the New World, while taking advantage of minicircle properties for ultra-sensitive PCR detection. Although the method has several limitations as a diagnostic tool, this pioneering work opens up avenues for the study of kDNA minicercles. In the context of important undergoing technological progress, high-throughput sequencing of kDNA minicircles could soon represent a useful tool for a variety of applications in Leishmania research.
SUPPLEMENTARY MATERIAL
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182017002013
Acknowledgments
We would like to thank Cécile Cassan for her help in the laboratory work. Leishmania strains were provided by the Biological Resource Center for Leishmania (Montpellier, France), the Parasitology and Mycology Laboratory of Cayenne Hospital (French Guiana), the Unit of Molecular Parasitology (Institute of Tropical Medicine, Antwerp, Belgium) and the Alexander von Humboldt Institute (Universidad Peruana Cayetano Heredia, Lima, Peru).
FINANCIAL SUPPORT
This work was supported by ‘Investissement d'Avenir’ grants managed by Agence Nationale de la Recherche (CEBA: ANR-10-LABX-25-01; TULIP: ANR-10-LABX-41, ANR-11-IDEX-0002-02; IAEL: ANR-06-SEST-20) as well as the Inkermann fund hosted at the Fondation de France.









