Introduction
Dourine or covering sickness is a sexually transmitted infection caused by Trypanosoma equiperdum, a haemoparasite that belongs to the Trypanozoon subgenus of Salivarian trypanosomes (Brun et al., Reference Brun, Hecker and Lun1998; Stevens and Brisse, Reference Stevens, Brisse, Maudlin, Holmes and Miles2004). This disease is characterized by genital inflammation, skin plaques and neurological signs, and mainly affects horses and other equids (Gizaw et al., Reference Gizaw, Megersa and Fayera2017).
The reversible phosphorylation of proteins is a ubiquitous mechanism of regulation that is vital for all living cells. In trypanosomatids, approximately 2% of their genome is composed of genes that encode for protein kinases, suggesting that protein phosphorylation plays a key role in the biology of these parasites (Naula et al., Reference Naula, Parsons and Mottram2005; Parsons et al., Reference Parsons, Worthey, Ward and Mottram2005). In the genomes of Trypanosoma brucei, Trypanosoma cruzi, Leishmania major and Trypanosoma evansi, there are genes that encode for three AGC protein kinases related to the catalytic subunits of the mammalian cAMP-dependent protein kinase (aka protein kinase A or PKA), and a gene homologous to the gene for the mammalian PKA regulatory subunits (http://tritrypdb.org). It has been suggested that these PKA catalytic subunit-like protein kinases might be involved in crucial cellular processes in trypanosomatids, such as osmoregulation, DNA repair, resistance to oxidative stress, activation of metabolic enzymes, replication, growth and cellular motility (Genestra et al., Reference Genestra, Cysne-Finkelstein and Leon2004; Parsons et al., Reference Parsons, Worthey, Ward and Mottram2005; Bao et al., Reference Bao, Weiss, Braunstein and Huang2008; Malki-Feldman and Jaffe, Reference Malki-Feldman and Jaffe2009). In this work, we partially purified and biochemically characterized a T. equiperdum protein kinase that recognizes fluorescently modified kemptide, which is a synthetic phosphate acceptor heptapeptide (sequence: LRRASLG) that resembles the local phosphorylation site sequence in pig liver pyruvate kinase and contains the consensus phosphorylation site for the PKA catalytic subunit (Kemp et al., Reference Kemp, Bylund, Huang and Krebs1975; Maller et al., Reference Maller, Kemp and Krebs1978; Kemp, Reference Kemp1980). Therefore, kemptide serves as a specific peptide substrate for PKA and PKA-like enzymes. Moreover, the parasite enzyme was capable of phosphorylating histone type II-AS, the synthetic peptide SP20 (sequence: TTYADFIASGRTGRRNSIHD), and the α isoform of the PKA type II regulatory subunit (RIIα-subunit), which are also known to function as substrates for the PKA catalytic subunit. The ATP:phosphotransferase activity of the T. equiperdum kemptide kinase was inhibited by both the PKA-specific heat-stable peptide inhibitor PKI-α and the synthetic peptide IP20 (sequence: TTYADFIASGRTGRRNAIHD), which is derived from PKI-α. However, the trypanosome kinase activity was not dependent on cAMP. Interestingly, the activity of this parasite enzyme was triggered upon nutritional stress driven by glucose starvation.
Materials and methods
Parasites
Cryopreserved T. equiperdum parasites from the Venezuelan TeAp-N/D1 strain were inoculated into Sprague–Dawley adult albino rats. The TeAp-N/D1 strain has been shown to be closely related to the T. equiperdum STIB841/OVI strain (Sánchez et al., Reference Sánchez, Perrone, Recchimuzzi, Cardozo, Biteau, Aso, Mijares, Baltz, Berthier, Balzano-Nogueira and Gonzatti2015). When more than 100 trypanosomes per microscopic field were detected in blood smears, the infected anaesthetized rats were bled via heart puncture using 2% EDTA (pH 8.0) as anticoagulant. Trypanosomes were purified by ion-exchange chromatography using a fibrous diethylaminoethyl (DEAE)-cellulose column equilibrated with phosphate-buffered saline (PBS) containing glucose (PBS-G) [57 mm Na2HPO4, 3 mm NaH2PO4, 43.8 mm NaCl and 1% glucose (pH 8.0)] (Lanham and Godfrey, Reference Lanham and Godfrey1970). Parasites eluting from the column were collected by centrifugation at 1475 g, for 10 min and washed three times with PBS-G buffer. The number of parasites was measured using a haemocytometer or Neubauer chamber.
Glucose deprivation of T. equiperdum parasites
Freshly purified parasites were incubated in the absence of glucose with PBS [57 mm Na2HPO4, 3 mm NaH2PO4 and 43.8 mm NaCl (pH 8.0)], for various times (0, 15, 30 and 60 min), at room temperature, under gentle and constant agitation. A sample of parasites that were deprived of glucose for 60 min was reversed by further incubation with glucose using the PBS-G buffer for 30 min. In all cases, trypanosomes were centrifuged at 1475 g, for 5 min, at 4 °C and the sedimented cells were stored at –80 °C until further use.
Kemptide kinase activity using an electrophoretic gel-shift assay
Kemptide kinase activity was measured using the Pep-Tag® non-radioactive method (Promega, Madison, WI, USA) with the fluorescently labelled kemptide (PepTag® A1 Peptide-LRRASLG) as substrate. Assays were performed as instructed by the manufacturer, and the reaction mixtures were separated by electrophoresis on a 1.2% agarose gel. When the commercial PepTag® A1 Peptide-LRRASLG component had run out of the kit, the rest of the experiments were performed with either the fluram-kemptide or the fluram-kemptide-Lys8 peptide (Araujo et al., Reference Araujo, Guevara, Lorenzo, Calabokis and Bubis2016) at a final concentration of 0.25 mg mL−1. Both kemptide and the kemptide-Lys8 peptide were chemically synthesized using the t-Boc methodology and were labelled with fluorescamine (fluram) as previously described (Araujo et al., Reference Araujo, Guevara, Lorenzo, Calabokis and Bubis2016). No preference for any of these fluorescently modified peptides was observed since all contained the specific substrate recognition site for the PKA catalytic subunit (sequence: RRXS/TΨ, where X is any amino acid and Ψ is a hydrophobic residue). Gels were revealed in a phototransilluminator (Fotodyne, Hartland, WI, USA) with UV light. The PKA catalytic subunit was purified from pig heart (Nelson and Taylor, Reference Nelson and Taylor1981) and used as a positive control.
Kemptide kinase activity using a high-performance liquid chromatography assay
ATP:phosphotransferase activity was also determined by using a high-performance liquid chromatography (HPLC) assay on a reversed-phase octadecylsilane column (Kemp, Reference Kemp1980; Luzi et al., Reference Luzi, Lyons, Peterson and Ellis2017). HPLC conditions were as described by Luzi et al. (Reference Luzi, Lyons, Peterson and Ellis2017), but the reaction was modified by employing fluram-kemptide as substrate. Phosphorylation reactions were initiated by incorporating the T. equiperdum protein kinase-containing fraction, and were terminated by the addition of 5% phosphoric acid. HPLC separations were carried out on a Waters 1525 binary pump system with a Symmetry® C18 column (3.9 × 150 mm, 5 µm, Waters, Milford, MA, USA), and the absorbance was monitored on a Waters 2487 dual wavelength detector. Since fluram-kemptide possesses a maximum excitation peak at ~400 nm (Araujo et al., Reference Araujo, Guevara, Lorenzo, Calabokis and Bubis2016), absorbance was measured at both 210 and 400 nm. Progress of the reactions was quantified by integrating the corresponding area peaks of the dephosphorylated substrate at 400 nm. The assay was initially standardized using the purified porcine heart PKA catalytic subunit (data not shown).
Partial purification of the T. equiperdum kemptide kinase
Freshly purified parasites (~109–1010 trypanosomes) were incubated in the absence of glucose for 1 h, as described above. Cells were extracted with lysis buffer [50 mm Tris-HCl (pH 8.0), 1 mm CaCl2, 1 mm MgCl2, 50 µ m phenyl methyl sulfonyl fluoride (PMSF), 1 mm benzamidine, and 10 µ m L-trans-epoxysuccinyl-leucylamido(4-guanidino)butane (E-64)] by sonication, using four cycles of 30 s each, with resting intervals of 2 min per cycle. The homogenate was centrifuged at 100 000 g for 1 h, at 4 °C, in order to obtain the corresponding parasite soluble fraction, which was loaded on a linear 5–20% sucrose gradient prepared in 50 mm Tris-HCl (pH 7.0), 0.1 mm EDTA, 0.15 M NH4Cl and 0.2 mm dithiothreitol. The sample was centrifuged at 100 000 g, for 20 h, at 5 °C. Fractions were collected from the top of the tube, and a sample of each fraction was assayed for kemptide kinase activity. Three peaks of enzymatic activity were obtained (see ‘Results’ section).
Fractions corresponding to peak 1 of the kemptide kinase activity were pooled and loaded on a 60 ml DEAE-Sepharose column that was previously equilibrated with 50 mm Tris-HCl (pH 7.0), 0.1 mm EDTA and 0.1 mm EGTA. Proteins were eluted from the column using a 0.01–1.0 M NaCl gradient in the same buffer containing 50 µ m PMSF, 1 mm benzamidine and 10 µ m E-64.
Fractions containing the kemptide kinase activity peak from the DEAE-Sepharose column were pooled and separated through a 90 mL Sephacryl S-300 gel-filtration column using 50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 3 mm Mg-acetate, 2 mm CaCl2, 5 mm β-mercaptoethanol, 50 µ m PMSF, 1 mm benzamidine and 10 µ m E-64 as elution buffer.
Effect of glucose on the T. equiperdum kemptide kinase enzymatic activities
Fresh purified T. equiperdum parasites were incubated in the absence of glucose with PBS, for 60 min, at room temperature. Glucose-deprived parasites were then incubated for 10 min with various concentrations of glucose (0–10 mm). Following homogenization of the parasite cells with lysis buffer, the resulting whole-cell extracts were centrifuged, and kemptide kinase activity was measured on the corresponding soluble fractions by HPLC (Luzi et al., Reference Luzi, Lyons, Peterson and Ellis2017).
Kemptide kinase activity was also determined on the partially purified trypanosome enzyme after incubation with different concentrations of glucose (0–1 mm).
Evaluation of potential substrates for the partially purified parasite protein kinase
The electrophoretic gel-shift assay was used to analyse whether the T. equiperdum kemptide kinase was capable of phosphorylating histone type II-AS from calf thymus (Sigma, Saint Louis, MO, USA) and a custom synthesized 20-residue peptide, SP20 (Life Tein, Somerset, NJ, USA), which is derived from the PKA-specific inhibitor PKI-α and possesses the following sequence: TTYADFIASGRTGRRNSIHD. In SP20, the Ala residue of PKI-α is substituted by a Ser at position 17. Kinase reactions were carried out as described by Lutz et al. (Reference Lutz, Pinon and Miller1994) using either 0.4 mg mL−1 of histone II-AS or 0.25 mg mL−1 of SP20.
Since the mammalian PKA RIIα-subunit contains a site that is autophosphorylated by the PKA catalytic subunit, the RIIα-subunit was purified from porcine heart (Taylor and Stafford, Reference Taylor and Stafford1978; Nelson and Taylor, Reference Nelson and Taylor1983) in order to explore its phosphorylation by the trypanosome protein kinase. Phosphorylation of the RIIα-subunit was performed as previously described (First et al., Reference First, Bubis and Taylor1988), with slight modifications. Briefly, purified RIIα-subunit from pig heart (5 µg) was incubated for 30 min, at room temperature, with an aliquot (15 µL) of the partially purified T. equiperdum kemptide kinase in the presence of 50 mm Tris-HCl (pH 7.5) containing 1 mm cAMP, 10 mm MgCl2 and 1 mm ATP. A control experiment was carried out in parallel using 1 µg of the purified PKA catalytic subunit from pig heart. Reactions were terminated by adding sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer (Laemmli, Reference Laemmli1970), and boiling the samples for 5 min. Then, the reaction mixtures were separated by SDS-PAGE on an 8% polyacrylamide slab gel and electrotransferred to nitrocellulose sheets (Towbin et al., Reference Towbin, Staehelin and Gordon1979). Membranes were first incubated with rabbit polyclonal anti-PKA RIIα-subunit antibodies (Santa Cruz Biotechnology Dallas, TX, USA, , dilution 1:5000), and then with alkaline phosphatase-conjugated antibodies against rabbit IgG (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA, dilution 1:1000). Polypeptide bands were visualized by the addition of 5-bromo-4-chloro-3 indolyl phosphate and nitro blue tetrazolium according to the supplier (Thermo Scientific Pierce, Waltham, MA, USA).
Biochemical characterization of the partially purified T. equiperdum kemptide kinase activity
Kemptide kinase activity was evaluated in the presence of 10 mm MgCl2 and 1 mm ATP at different temperatures (−20 to 60 °C) and pH (2.0–11.0), to establish the optimal conditions for enzymatic activity. The optimal pH was determined by using suitable buffers (100 mm), which were selected based on their pKa: maleate (pH 2.0), glycine (pH 3.0), formate (pH 4.0), acetate (pH 5.0), 2-(N-morpholino)ethanesulfonic acid (pH 6.0), [3-(N-morpholino)propanesulfonic acid] (pH 7.0), Tris (pH 8.0) and N-cyclohexyl-3-aminopropanesulfonic acid (pH 9.0–11.0). In order to confirm that the effect on the enzymatic activity was produced by changes in the pH rather than to variations in the buffer system, we used a series of buffers (100 mm) with overlapping effective pH ranges, namely citrate (pH 2.0, 3.0, 4.0, 5.0 and 6.0), phosphate (pH 6.0, 7.0 and 8.0), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, pH 7.0 and 8.0), Tris (pH 7.0, 8.0 and 9.0) and carbonate (pH 8.5, 9.0, 10.0 and 11.0).
The effect on the trypanosome kinase activity of cAMP and the recombinant isoform α of the PKA heat-stable inhibitor PKI from rabbit (PKI-α, Calbiochem, San Diego, CA, USA) was also evaluated. We choose two concentrations of cAMP, 5 µ m that lied in the expected physiological range, and 5 mm that was extremely high and non-physiological. The inhibitory effect of PKI-α was measured by using 0.9, 9 and 36 units of the PKA-specific inhibitor peptide. The inhibitory effect of PKI-α was corroborated by using a custom-synthesized 20-amino acid peptide, IP20 (sequence: TTYADFIASGRTGRRNAIHD, Life Tein, Somerset, NJ, USA), which is derived from PKI-α and contains its inhibitory motif. The partially purified protein kinase from T. equiperdum was pre-incubated with IP20 (0.25 mg mL−1) in 50 mm Tris-HCl (pH 7.5), for 30 min at 4 °C, and then the remainder components of the reaction mixture were added (0.25 mg mL−1 fluram-kemptide, 10 mm MgCl2 and 1 mm ATP). Kemptide kinase activity was monitored by using the electrophoretic gel-shift assay.
The specificity of nucleotide triphosphates and divalent cations was monitored by evaluating the enzymatic activity using 1 mm ATP, GTP or CTP (in the presence of 10 mm MgCl2); and 10 mm MgCl2, ZnCl2, MnCl2, CoCl2, CuCl, CaCl2 or FeSO4 (in the presence of 1 mm ATP), respectively.
The inhibitory effect of AMP, ADP, GMP, GDP, GTP, CTP, β,γ-imido-adenosine 5′-triphosphate (AMP-PNP) and 5′-[p-(fluoro sulfonyl)benzoyl] adenosine (FSBA) on the parasite enzymatic activity was measured by pre-incubating the partially purified kemptide kinase with 2 mm of each compound for 30 min, under ice, in the presence of 10 mm MgCl2. Subsequently, the kinase assay was performed as described above following incubation with 1 mm ATP for 1 h.
To determine the inhibitory effect of the different divalent cations on the parasite enzymatic activity, the partially purified trypanosome kemptide kinase was pre-incubated with 20 mm ZnCl2, MnCl2, CoCl2, CuCl, CaCl2 or FeSO4, for 30 min, under ice, in the presence of 1 mm ATP. Then, the kinase assay was assayed as described above following incubation with 10 mm MgCl2 for 1 h.
Determination of the Km for ATP and kemptide of the T. equiperdum kemptide kinase
The K m values for ATP and kemptide of the partially purified parasite enzyme were determined by HPLC (Luzi et al., Reference Luzi, Lyons, Peterson and Ellis2017). To measure the K m for ATP, the reactions were performed in 50 mm Tris-HCl (pH 7.4), 10 mm MgCl2, 30 µ m fluram-kemptide and various concentrations of ATP (0–70 µ m). To measure the K m for kemptide, the mixtures contained 50 mm Tris-HCl (pH 7.4), 10 mm MgCl2, 1 mm ATP and various concentrations of fluram-kemptide (0–50 µ m). Three independent experiments were performed for each concentration, and K m values were estimated by regression analysis of double reciprocal plots (Lineweaver and Burk, Reference Lineweaver and Burk1934). Values were reported as the mean ± the standard error of the mean (s.e.m.).
SDS-PAGE
Fractions obtained from all purification steps were separated by SDS-PAGE according to Laemmli (Reference Laemmli1970), and revealed using silver staining.
Results
Activation of kemptide kinase activities by glucose deprivation of T. equiperdum parasites
By using the electrophoretic gel-shift assay, no kemptide kinase activities were detected in whole-cell homogenates, particulate fractions or clarified soluble fractions from trypanosomes purified in the presence of 1% glucose, which corresponds to 55.5 mm (Fig. 1A). Equivalent results were obtained in the absence or presence of either 5 µM or 5 mM cAMP. However, an increase in kemptide phosphorylation was achieved in homogenates and soluble fractions from parasites that underwent glucose limitation for 15, 30 and 60 min (Fig. 1B). No kemptide kinase activities were observed in the particulate fraction from glucose-starved T. equiperdum microbes revealing that when activated, these enzymatic activities were exclusively localized in the coresponding soluble fraction (Fig. 1B). Similar results were found by the HPLC assay when kemptide kinase activities were determined on the clarified soluble fractions from glucose-deprived T. equiperdum parasites (Fig. 2A). Moreover, the activation of kemptide kinase activities elicited in glucose-deprived parasites was readily reversed, and these enzymes were inhibited upon re-addition of 1% glucose for 30 min to parasites that experienced glucose fasting for 60 min (Fig. 1B, 60:30). As also shown in Fig. 1B, no stimulation by exogenously added cAMP (5 µ m) was obtained in these experiments. Similar results were found when 5 mm cAMP was used (data not shown). If anything, a decrease in the phosphorylation signal of the fluorescently labelled kemptide substrate was observed when cAMP was present, suggesting that cAMP might be inhibiting the parasite kemptide kinase activities. Although the activity of the homogenate sample at the 60 min time point appears to be slightly higher upon addition of cAMP than in the absence of cAMP (Fig. 1B), we feel that this particular result is an artefact produced by the qualitative features of the electrophoretic gel-shift assay. Absence of stimulation by cAMP was consistent in many independent experiments that were carried out. On the basis of how PKA enzymes are commonly defined, these results established that the parasite kemptide kinase activities represent cAMP-independent enzymes.

Fig. 1. Effect of glucose deprivation on the parasite kemptide kinase activities by using the electrophoretic gel-shift assay. (A) No kinase activities were detected when trypanosomes were purified in the presence of glucose. T. equiperdum parasites were purified through a DEAE-cellulose column using PBS-G (PBS containing 1% glucose), and the purified trypanosomes were immediately used (0 min). Enzymatic activities were qualitatively evaluated on the parasite whole-cell homogenate (H), particulate fraction (P) and clarified soluble fraction (S) by means of the Promega Pep-Tag® non-radioactive assay, using the PepTag® A1 kemptide as substrate. Determinations were made in the absence (−) or presence of either 5 µ m or 5 mm cAMP (+). (B) Glucose starvation activates kemptide kinase activities from T. equiperdum. Freshly purified parasites were incubated with PBS lacking glucose for 15, 30 and 60 min (GD = glucose deprivation). Also, a sample of the parasites deprived of glucose for 60 min was re-incubated with PBS-G for 30 min (60:30). Kemptide kinase activities were estimated on the homogenates (H), particulate fractions (P) and clarified soluble fractions (S) of all trypanosome samples by using the Promega Pep-Tag® non-radioactive assay. Measurements were made in the absence (−) or presence of 5 µ m cAMP (+). In (A) and (B), gels were revealed with UV light in a phototransilluminator. The arrowhead and arrow indicate the non-phosphorylated and phosphorylated fluorescently labelled peptide, respectively. Purified PKA catalytic subunit from porcine heart was employed as a positive control (C+). The reaction mixture without any added enzyme was used as a negative control (C−).

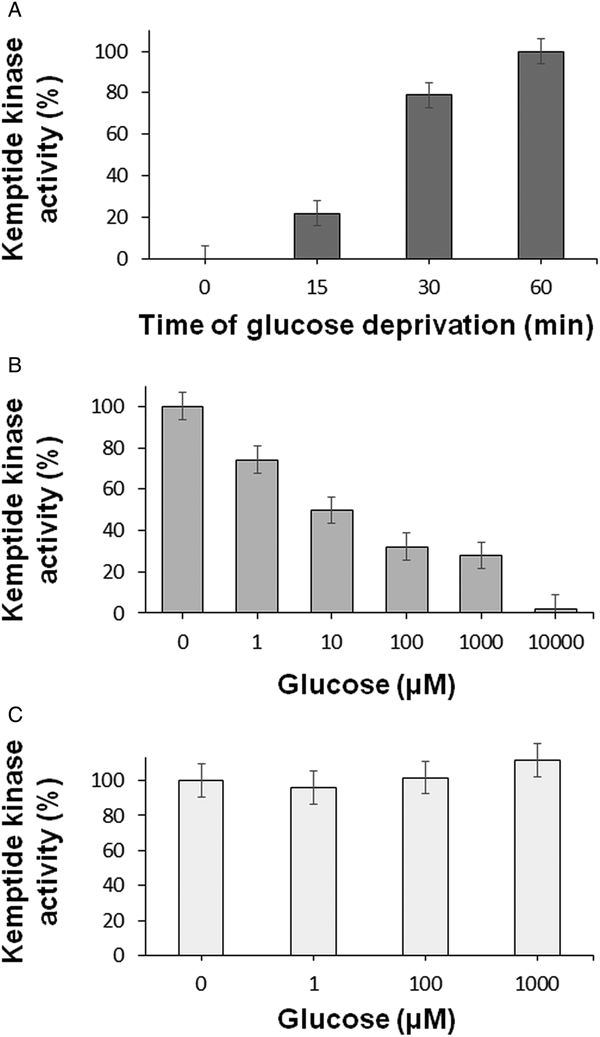
Fig. 2. Effect of glucose deprivation on the T. equiperdum kemptide kinase activities by using the HPLC assay. (A) Time course of the effect of glucose fasting. Fresh purified parasites were incubated in the absence of glucose with PBS for various times (0, 15, 30 and 60 min). Parasites were then extracted with lysis buffer and the homogenates were centrifuged to obtain the corresponding clarified soluble fractions. Kemptide kinase activities were assayed by HPLC. (B) Incubation of T. equiperdum parasites with different concentrations of glucose. Freshly purified trypanosomes were deprived of glucose for 60 min, and then were incubated for 10 min with increasing concentrations of glucose (0–10 mm). Following homogenization and centrifugation, kemptide kinase activities were evaluated on the corresponding soluble fractions by HPLC. (C) Incubation of the partially purified kemptide kinase from T. equiperdum with different concentrations of glucose. The enriched kemptide kinase-containing fraction that was obtained as described in Fig. 3 was incubated with various concentrations of glucose (0–1 mm). Kemptide kinase activities were measured by HPLC. In all cases, values were reported as the mean ± s.e.m.
Nutritionally stressed T. equiperdum parasites were incubated with increasing amounts of glucose in order to determine its half maximal inhibitory concentration (IC50). Following homogenization and centrifugation, kemptide kinase activities were evaluated on the corresponding soluble fractions by HPLC. As seen in Fig. 2B, a concentration of ~10 µ m glucose was required for a 50% inactivation of these enzymes. These results showed that the inhibition by glucose of the parasite kinase activities was concentration dependent.
Partial purification of a T. equiperdum kemptide kinase activity
Freshly isolated T. equiperdum parasites were incubated in the absence of glucose for 60 min in order to stimulate the trypanosome kemptide kinase activities. Parasites were homogenized by sonication and centrifuged to separate the clarified soluble fraction, which contains the kemptide kinase activities, from the particulate fraction, and the soluble fraction was fractionated by sedimentation throughout a 5–20% sucrose gradient. Three peaks of kemptide kinase activity were detected (Fig. 3A): fractions 4–10 (peak 1), fractions 14–18 (peak 2) and fractions 20–22 (peak 3). We selected peak 1 for further purification.

Fig. 3. Partial purification of a protein kemptide kinase from T. equiperdum. (A) Sedimentation by ultracentrifugation on a sucrose gradient. The soluble fraction from glucose-starved T. equiperdum was subjected to sedimentation by ultracentrifugation on a 5–20% sucrose gradient. Fractions were collected from the top of the tube and assayed for kinase activity employing the fluram-kemptide-Lys8 peptide as a substrate. Purified PKA catalytic subunit from pig heart was used as a positive control (C+). The reaction mixture without any added enzyme was used as a negative control (C−). (B) Separation by anion-exchange chromatography. Fractions 4–10 from the sucrose gradient were pooled and separated on a DEAE-Sepharose column. The chromatogram (top) illustrates the protein profile (absorbance at 280 nm) and the ionic strength of the eluting fractions, which were assayed for kinase activity using the fluram-kemptide-Lys8 peptide as a substrate. The bottom panel shows the fractions containing the kemptide kinase peak, which is highlighted with an asterisk (*) in the chromatogram. (C) Separation by gel-filtration chromatography. Fractions 26–30 from the DEAE-Sepharose column were collected, concentrated and loaded on a Sephacryl S-300 column. Plotted is the absorbance at 280 nm against the elution volume of the fractions (top), which were assayed for kinase activity using the fluram-kemptide-Lys8 peptide as a substrate. The bottom panel illustrates the elution volume of the fractions containing the kemptide kinase peak, which is highlighted with an asterisk (*) in the chromatogram. In all cases, the arrowhead and arrow indicate the non-phosphorylated and phosphorylated fluorescently labelled peptide, respectively.
Peak 1 from the sucrose gradient was separated by anion exchange chromatography through a DEAE-Sepharose column. Proteins were eluted by increasing the ionic strength of the washing buffer with NaCl and kemptide kinase activity was detected between fractions 26 and 30 (Fig. 3B). This kinase peak coincided with the major absorbing peak in the protein profile, which eluted at a concentration of 250 mm NaCl (Fig. 3B). Fractions 26–30 from the DEAE-Sepharose column were collected, concentrated and loaded onto a Sephacryl S-300 size exclusion chromatography column. Proteins were eluted from the column, and a peak of kemptide kinase activity was detected between elution volumes of 85.5 and 86.0 mL, with its maximum at 85.75 mL (Fig. 3C).
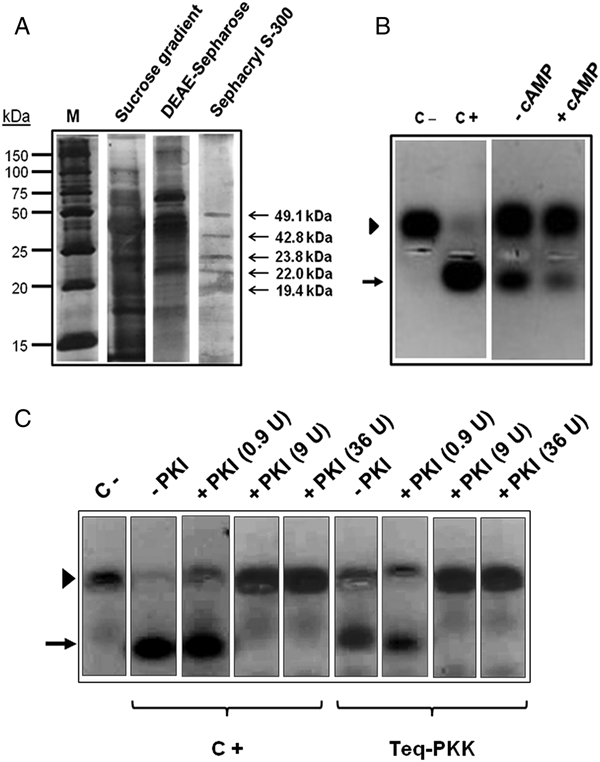
The purity of the kemptide kinase activity peaks from the sucrose gradient, the DEAE-Sepharose chromatography and the gel filtration column, was analysed by SDS-PAGE using a 12% polyacrylamide gel (Fig. 4A). As shown in the figure, five major polypeptide bands of 49.1, 42.8, 23.8, 22.0 and 19.4 kDa were observed in the polyacrylamide gel after the three purification steps, revealing that the T. equiperdum kemptide kinase enzyme was only partially purified. This enriched kemptide kinase-containing fraction was named ppTeq-PKK for ‘partially purified T. equiperdum protein kemptide kinase’ and was employed in all further assays performed to characterize the enzyme.

Fig. 4. (A) Separation by SDS-PAGE of the T. equiperdum kemptide kinase purification steps. Proteins were revealed by silver staining. Arrows indicate the molecular masses of the polypeptide bands contained in the kemptide kinase-enriched fraction. M = molecular weight markers. (B) ppTeq-PKK is a cAMP-independent kemptide kinase. Kinase activity was measured by the Promega Pep-Tag® non-radioactive assay, using the PepTag® A1 kemptide as substrate. Determinations were made in the absence (−) or presence of 5 µ m cAMP (+). (C) PKI-α inhibits ppTeq-PKK. ppTeq-PKK (Teq-PKK) was incubated with various units (U) of PKI-α (0.9, 9 and 36 units). Kinase activity was assayed using the fluram-kemptide-Lys8 peptide as substrate. In (B) and (C), the arrowhead and arrow indicate the non-phosphorylated and phosphorylated peptide, respectively. Purified PKA catalytic subunit from pig heart was utilized as a positive control (C+). The reaction mixture without any added enzyme was used as a negative control (C−).
As illustrated in Fig. 4B, and agreeing with the results shown in Fig. 1B, no stimulation by exogenously added cAMP was acquired on ppTeq-PKK, which confirmed that the trypanosome kinase corresponds to a cAMP-independent enzyme. As previously seen in Fig. 1B, a decrease in the phosphorylation signal of the fluorescently labelled kemptide was observed when cAMP was present (Fig. 4B), which again suggests that cAMP might be inhibiting the enzymatic activity perhaps by competing with ATP for the ATP-binding site on the parasite protein kinase.
Interestingly, the recombinant PKI-α from rabbit, which is a 77-amino acid protein that is a potent and highly specific inhibitor of PKA catalytic subunits, inhibited similarly both ppTeq-PKK and the purified PKA catalytic subunit from pig heart (Fig. 4C). Since PKI-α is not known to inhibit any other type of protein kinase, ppTeq-PKK appears to be a PKA catalytic subunit-like protein. This result was corroborated by using IP20, a peptide that contains the inhibitory sequence of PKI-α. As seen in Fig. S1, IP20 also inhibited both the purified PKA catalytic subunit from porcine heart (lane C+, IP20) and ppTeq-PKK (lane Teq-PKK, IP20).
With the intention of evaluating whether glucose inhibition was caused by a direct interaction between the monosaccharide and the enzyme, ppTeq-PKK was incubated with various concentrations of glucose. As shown in Fig. 2C, glucose did not inactivate the parasite protein kinase, implying that the sugar is not affecting directly the trypanosome enzyme but an upstream component of this kemptide kinase signalling pathway. These findings were consistent with our previous results that showed kemptide kinase activities following fractionation by sedimentation throughout a 5–20% sucrose gradient. Although the centrifuged fractions contained sucrose, three peaks of kemptide kinase activity were identified (Fig. 3A).
Evaluation of other substrates
Although kemptide has been shown to be a specific peptide substrate for PKA and PKA-like enzymes, it is also known that it can be phosphorylated by other protein kinases (MacAla et al., Reference MacAla, Hayslett and Smallwood1998). For that reason, we used the electrophoretic gel-shift assay to determine whether ppTeq-PKK was capable of phosphorylating two additional substrates, histone type II-AS and SP20, which are also known to serve as substrates for PKA and PKA-like enzymes. Analogous to kemptide, both histone type II-AS and SP20 were phosphorylated by the trypanosome enzymatic activity (Fig. 5A).

Fig. 5. Phosphorylation of histone type II-AS, SP20 and the PKA RIIα-subunit by ppTeq-PKK. (A) The trypanosome enzyme was capable of phosphorylating kemptide, histone type II-AS and SP20 by means of the electrophoretic gel-shift assay. Kinase reactions were performed in the absence (−) or presence (+) of ppTeq-PKK. The arrowhead and arrow indicate the migration of the non-phosphorylated and phosphorylated substrates, respectively. (B) Purified RIIα-subunit alone (RII) or in the presence of either the pig heart PKA catalytic subunit (RII + C) or ppTeq-PKK (RII + Teq-PKK) was incubated with 1 mm cAMP, 10 mm MgCl2 and 1 mm ATP in 50 mm Tris-HCl (pH 7.5). Following incubation for 30 min, at room temperature, the reaction mixtures were separated by SDS-PAGE on an 8% polyacrylamide slab gel and electrotransferred to nitrocellulose. The blot was revealed by using anti-RIIα-subunit antibodies. Arrows indicate the migration and apparent molecular masses of the dephosphorylated (RII) and phosphorylated (RII-P) forms of the monomeric RIIα-subunit. An endogenous proteolytic fragment of the RIIα-subunit is also shown (COOH-RII).
The PKA RIIα-subunit contains a phosphorylatable epitope within the catalytic subunit inhibitory sequence. As previously shown (First et al., Reference First, Bubis and Taylor1988), SDS-PAGE distinguishes the phospho and dephospho forms of the pig heart RIIα-subunit readily. Immunoblotting using anti-RIIα-subunit antibodies illustrated that upon phosphorylation by the PKA catalytic subunit, the purified RIIα-subunit from pig heart changed its electrophoretic mobility on SDS-polyacrylamide gels from an apparent molecular mass of 55 kDa to an apparent molecular mass of 57 kDa (Fig. 5B, lane RII + C). Interestingly, ppTeq-PKK was also capable of phosphorylating the purified RIIα-subunit from porcine cardiac muscle, as shown by the appearance of a doublet of 55 and 57 kDa (Fig. 5B, lane RII + Teq-PKK). It is also known that PKA regulatory subunits possess a hinge region that is very susceptible to proteolysis and is located adjacent to the catalytic subunit inhibitory sequence. Limited proteolysis cleaves the regulatory subunits into carboxy-terminal fragments that retain the cAMP-binding properties of the native proteins but that have lost the primary contacts between the two subunits. These carboxy-terminal fragments represent about the last two-thirds of the proteins. Figure 5B also shows a band that migrates with an apparent molecular mass of ~37 kDa, and is recognized by the anti-RIIα-subunit antibodies. The 37 kDa polypeptide band must correspond to the RIIα-subunit proteolytic fragment (COOH-RII) that is formed by endogenous proteolysis (Bubis and Taylor, Reference Bubis and Taylor1987).
Determination of the molecular weight and hydrodynamic parameters of the kemptide kinase from T. equiperdum
ppTeq-PKK was applied to a size-exclusion column that was calibrated using markers with known molecular masses. By plotting the partition coefficient (Kav) of each standard vs the logarithm of its molecular weight (Laurent and Killander, Reference Laurent and Killander1964), a molecular mass of 39.07 kDa was determined for the partially purified trypanosome enzyme (Fig. S2A). Additionally, a linear relationship was obtained by plotting (−log Kav)1/2 against the Stokes radius of each standard protein (Siegel and Monty, Reference Siegel and Monty1966), and a Stokes radius of 2.74 nm was calculated for the partially purified parasite protein (Fig. S2B). A sedimentation coefficient of 4.06 S was measured for the parasite kemptide kinase by subjecting the enzyme to ultracentrifugation in the presence of a mixture of proteins with known sedimentation coefficients (Fig. S3A). As seen in Fig. S3B, a molecular mass of 51.73 kDa was obtained by plotting the sedimentation coefficient against the (molecular weight)2/3 of each standard (Martin and Ames, Reference Martin and Ames1961). For spherical molecules, the molecular weight of a species can also be calculated from a combination of the measured Stokes radius and sedimentation coefficient (Siegel and Monty, Reference Siegel and Monty1966; Johansson et al., Reference Johansson, Majka and Burgers2001). Using this approximation, a molecular mass of 43.55 kDa was estimated for the parasite enzyme (Fig. S3C). Combining all these results, the native size of ppTeq-PKK appears to be between 39.07 and 51.73 kDa. As shown above in Fig. 4A, SDS-PAGE revealed polypeptide bands with apparent molecular masses of 42.8 and 49.1 kDa. Therefore, the parasite enzyme might correspond to one of these bands.
The frictional coefficient f/f o can be determined from the molecular weight and the Stokes radius (Bloomfield et al., Reference Bloomfield, Dalton and van Holde1967). The calculated frictional ratio value for the partially purified kemptide kinase was 1.22, 1.24 or 1.25 when values of the molecular mass obtained either by gel filtration (39.07 kDa), or by the combination of the measured Stokes radius and sedimentation coefficient (43.55 kDa), or by sedimentation (51.73 kDa) were employed, respectively. These findings indicate that the parasite enzyme is a globular and compact protein (Erickson, Reference Erickson2009). Since the frictional coefficient not only contains information about the molecular shape of the protein but also about the hydration effects, an upper boundary on the contributions of hydration can be assessed presuming that all deviations of f/f o from unity are due to hydration (Darling et al., Reference Darling, Holt and Ackers2000). The maximal value of hydration for the trypanosome kemptide kinase was 0.57–0.67 g of H2O per gram of protein (depending on the experimental value used for f/f o), which slightly diverges from the 0.3–0.5 g of H2O per gram value of a typical medium-sized and compact protein. This result also suggests that the T. equiperdum enzyme is globular and compact.
We were not able of determine the hydrodynamic parameters of the trypanosome kemptide kinase activities that fractionated with peaks 2 and 3 following sedimentation by ultracentrifugation (Fig. 3A). The migration volumes of these fractions were higher than that of alcohol dehydrogenase, which was the largest of the protein standards that were used for calibration. Therefore, the parasite kemptide kinase activities in peaks 2 and 3 possessed molecular masses higher than 150 kDa, and their sedimentation coefficients fell outside the linear range of the curve shown in Fig. S3A. Such high sizes for these protein kinases are suggestive of potential formation of complexes with other interacting proteins.
Biochemical characterization of the T. equiperdum kemptide kinase
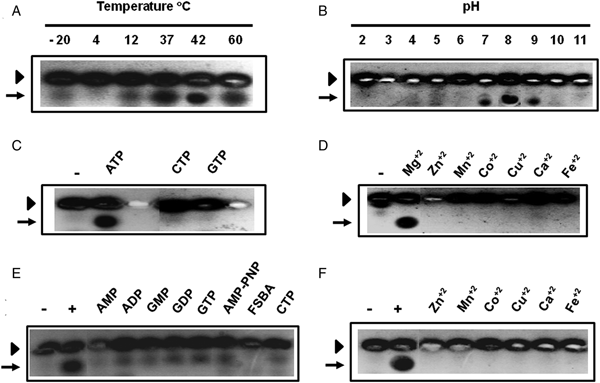
Kemptide kinase activity was assayed at different temperatures and pH, showing optimal performance at 37 °C (Fig. 6A) and pH 8.0 (Fig. 6B). Since two variables, the pH and the buffer system, were changed in Fig. 6B, we carried out an additional experiment that incorporated chosen buffer systems on the basis of their overlapping effective pH ranges (Fig. S4). Although slight effects caused by the buffer system on the trypanosome enzymatic activity were noticed, these results ratified that pH 8.0 is the optimal pH for ppTeq-PKK.

Fig. 6. Characterization of ppTeq-PKK. Measurement of the optimal temperature (A) and pH (B). Determination of the specificity for nucleotide triphosphates (C) and divalent cations (D). In (C) and (D), the reaction mixture without any added enzyme was used as a negative control (−). (E) Inhibitory effect of nucleotides and analogues of ATP in the presence of Mg2+. Samples containing the parasite enzyme were pre-incubated with 2 mm of each compound, and then assayed using 1 mm ATP. Control experiments were performed in the absence (−) and presence (+) of ATP. (F) Inhibitory effect of divalent cations in the presence of ATP. Samples containing the T. equiperdum kinase were pre-incubated with 20 mm of each divalent cation, and then assayed using 10 mm Mg2+. Control experiments were performed in the absence (−) and presence (+) of Mg2+. In all cases, kinase activity was assayed employing the fluram-kemptide-Lys8 peptide as a substrate. The arrowhead and arrow indicate the non-phosphorylated and phosphorylated peptide, respectively.
The parasite kinase activity was only detected in the presence of ATP (Fig. 6C) and Mg2+ (Fig. 6D), and no activity was perceived neither when GTP or CTP, nor when other divalent cations, such as Zn2+, Mn2+, Co2+, Cu2+, Ca2+ and Fe2+ were employed as co-substrates (Fig. 6C and D). However, the enzymatic activity of the parasite kemptide kinase was inhibited by other nucleotides and/or analogues of ATP, such as AMP, ADP, GMP, GDP, GTP, AMP-PNP, FSBA and CTP, and by other divalent cations (Zn2+, Mn2+, Co2+, Cu2+, Ca2+ and Fe2+) (Fig. 6E and F). Since the trypanosome enzyme was inhibited by such a variety of nucleotides, the reduction in kinase activity that was previously seen in the presence of cAMP (Figs 1B and 4B) must be caused by inhibition rather than by a direct influence on the enzymatic activity. As the other nucleotides, cAMP is probably binding and competing with ATP for the protein kinase ATP-binding site.
Determination of the K m for ATP and kemptide of the T. equiperdum kemptide kinase
Kinase activity was assayed by the HPLC methodology described by Luzi et al. (Reference Luzi, Lyons, Peterson and Ellis2017), which was modified by using fluram-kemptide as substrate. Typical Michaelis–Menten curves for ATP and fluram-kemptide were obtained for the trypanosome enzyme (Fig. 7). K m values for ATP and kemptide of 11.8 ± 4.1 µ m (Fig. 7A) and 24.7 ± 3.8 µ m (Fig. 7B), respectively, were determined for the parasite kinase after plotting the data on double reciprocal Lineweaver–Burk plots.

Fig. 7. Determination of the K m for ATP (A) and kemptide (B) of ppTeq-PKK. (A) The analysis of the phosphorylation of fluram-kemptide was assayed by HPLC. To determine the K m for ATP, various concentrations of ATP (0–70 µ m) were employed with a fixed concentration of fluram-kemptide (30 µ m). To measure the K m for kemptide, various concentrations of fluram-kemptide (0–50 µ m) were used with a fixed concentration of ATP (1 mm). In (A) and (B), the left and right panels represent the Michaelis–Menten curves and the Lineweaver–Burk double-reciprocal plots, respectively. K m values were reported as the mean ± s.e.m.
Discussion
Here, we report a novel protein kinase that was partially purified from the Venezuelan TeAp-N/D1 isolate of T. equiperdum. This enzyme was capable of recognizing kemptide, histone II-AS, SP20 and the PKA RIIα-subunit, which are specific substrates for the PKA catalytic subunit, and was inhibited by PKI-α and IP20, which are specific peptide inhibitors for the PKA catalytic subunit, but surprisingly, it was not stimulated by cAMP. Noticeably, the T. equiperdum kinase was activated when parasites were subjected to nutritional restriction caused by lack of glucose in the medium.
Glucose is the leading source of energy in the majority of eukaryotes, being glycolysis and mitochondrial respiration the sequential glucose oxidation routes. Many trypanosomatids live in carbohydrate-rich habitats inside their hosts. For instance, T. equiperdum, as well as T. evansi and the trypomastigote slender and stumpy stages of T. brucei, live in the bloodstream of their mammalian host, which warrants a permanent source of glucose. Parasite trypanosomes living in carbohydrate-rich environments seem to depend on carbohydrates for their energy metabolism, and do not require a cytochrome-containing respiratory chain or an elaborate mitochondrial metabolism (Tielens and van Hellemond, Reference Tielens and van Hellemond2009). Since glucose deprivation activates the T. equiperdum protein kinase described here, this enzyme may perhaps participate in a signalling cascade triggered by nutritional and/or energy stress. This metabolic signalling network might be implicated in controlling parasite survival during glucose fasting.
In most organisms, the PKA holoenzyme is an inactive tetrameric complex composed of two catalytic subunits bound to a dimer of regulatory subunits. Binding of cAMP to the regulatory subunits leads to the dissociation and activation of the two catalytically active catalytic subunit monomers (Taylor, Reference Taylor1989). Analysis of the complement of protein kinases in the completed genomes of various trypanosomatids have indicated that there are genes that encode for protein kinases related to the PKA catalytic subunits from higher eukaryotes (http://tritrypdb.org). The first draft of the T. equiperdum genome (Hébert et al., Reference Hébert, Moumen, Madeline, Steinbiss, Lakhdar, Van Reet, Büscher, Laugier, Cauchard and Petry2017), also described the presence of two protein kinases that are homologous to the PKA catalytic subunits [NCBI GenBank IDs: SCU68910.1 and SCU64246.1 (https://www.ncbi.nlm.nih.gov/genbank/)]. A gene homologous to the gene for the mammalian PKA regulatory subunits has also been reported in trypanosomatid parasites (http://tritrypdb.org); and recently, we have identified a 57 kDa polypeptide band corresponding to the PKA regulatory subunit-like protein from the T. equiperdum TeAp-N/D1 isolate (Calabokis et al., Reference Calabokis, González, Merchán, Escalona, Araujo, Sanz-Rodríguez, Cywiak, Spencer, Martínez and Bubis2016). Antibody cross-reactivity clearly established the high level of conservation exhibited between the T. equiperdum PKA regulatory subunit-like protein and its mammalian counterparts; however, the trypanosome protein differed from other PKA regulatory subunits since it is a monomer that is not capable of binding cAMP (Bubis et al., Reference Bubis, Martínez, Calabokis, Ferreira, Sanz-Rodríguez, Navas, Escalona, Guo and Taylor2018). It is remarkable to find that the parasite kinase enzymatic activity is inhibited or activated depending on the level of glucose in the incubation medium, which is reminiscent of the occurrence of an inactive holoenzyme, or another enzymatic form that is hindered by some inhibitory or regulatory molecule under high-glucose conditions. Yet, preliminary results from our laboratory using co-immunoprecipitation and pull-down experiments have also shown that there is no formation of a holoenzyme type of complex between the T. equiperdum PKA regulatory subunit-like protein and the kemptide kinase partially purified here (data not shown). These early results suggest that another inhibitory agent is responsible for the regulation of the kemptide kinase activity when glucose is not restricted.
Free PKA catalytic subunits from mammals have K ms for ATP and kemptide of ~10–20 µ m (Smith et al., Reference Smith, Radzio-Andzelm, Madhusudan, Akamine and Taylor1999; Moore et al., Reference Moore, Adams and Taylor2003; Mena-Ulecia et al., Reference Mena-Ulecia, Vergara-Jaque, Poblete, Tiznado and Caballero2014; Luzi et al., Reference Luzi, Lyons, Peterson and Ellis2017), which are similar to the K m values determined here for ppTeq-PKK. Moreover, the molecular mass and hydrodynamic properties found for the parasite kemptide kinase were comparable to those reported for the mammalian PKA catalytic subunits (Sugden et al., Reference Sugden, Holladay, Reimann and Corbin1976; Rubin et al., Reference Rubin, Rangel-Aldao, Sarkar, Erlichman and Fleischer1979; Zoller et al., Reference Zoller, Kerlavage and Taylor1979), and for PKA-like enzymes from other trypanosomatids (Banerjee and Sarkar, Reference Banerjee and Sarkar1992; Ochatt et al., Reference Ochatt, Ulloa, Torres and Téllez-Iñón1993). All these results indicate that the T. equiperdum enzyme is a globular protein as is the free PKA catalytic subunit from mammals, and suggest that the parasite protein is probably monomeric. On the basis of their calculated molecular weight, the gene product of either the reported SCU68910.1 or SCU64246.1 genes from T. equiperdum (https://www.ncbi.nlm.nih.gov/genbank/) might correspond to the kemptide kinase characterized here. Bioinformatics analyses using the Clustal Omega program (http://www.ebi.ac.uk/Tools/msa/clustalo/) of the sequences of the two T. equiperdum protein kinases, the three protein kinases identified in T. evansi [TevSTIB805.9.7920, TevSTIB805.9.7870 and TevSTIB805.10.13650 (http://tritrypdb.org)] and the α isoform of the Mus musculus PKA catalytic subunit (GenBank ID: AAH54834.1, https://www.ncbi.nlm.nih.gov/genbank/), yielded a 47–51% identity between the parasite kinases and the mouse protein, and an expected identity of >97% between the T. equiperdum and T. evansi protein kinases (data not shown).
If the parasite kinase corresponds to a PKA type of enzyme, it is unexpected to find that cAMP is not required for its activation. However, there are several reports describing cAMP-independent activation of PKA in other systems (Zhong et al., Reference Zhong, SuYang, Erdjument-Bromage, Tempst and Ghosh1997; Dulin et al., Reference Dulin, Niu, Browning, Ye and Voyno-Yasenetskaya2001; Ferraris et al., Reference Ferraris, Persaud, Williams, Chen and Burg2002; Zhang et al., Reference Zhang, Duan, Binkley, Li, Uhler, Logsdon and Simeone2004; Yang et al., Reference Yang, Lee, Zhang, Sans and Simeone2008; Kohr et al., Reference Kohr, Traynham, Roof, Davis and Ziolo2010). Even in Leishmania donovani that is another trypanosomatid parasite, Banerjee and Sarkar (Reference Banerjee and Sarkar1992) have reported the characterization of a cyclic nucleotide-independent PKA enzyme that recognized kemptide as a substrate. Trypanosoma equiperdum parasites may use similar strategies to activate this PKA-like protein in a cAMP-independent manner.
Control of diseases caused by trypanosomatid parasites relies mostly on chemotherapy. However, there is a very narrow battery of drugs and they generally have limitation issues, such as high toxicity and emerging resistance. Accordingly, finding new drug targets is an important goal in order to develop novel anti-parasitic chemotherapeutic agents. Since phosphorylation seems to play a key role in trypanosomatid parasite biology (Parsons et al., Reference Parsons, Worthey, Ward and Mottram2005), and given that protein kinases are involved in regulating cell cycle control, proliferation, differentiation and response to stress, these enzymes represent promising drug targets for diseases caused by parasitic protozoa (Naula et al., Reference Naula, Parsons and Mottram2005). Although the physiological role of the T. equiperdum kemptide kinase characterized here is yet to be found, this enzyme might be an attractive drug target. The findings presented here open new avenues to study signal transduction pathways in T. equiperdum, and is a promising starting point that may eventually lead to the development of new anti-trypanosomosis therapeutics for treatment.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182018001920.
Author ORCIDs
José Bubis http://orcid.org/0000-0002-8839-6745.
Acknowledgements
We thank Oscar Noya and María A. Lorenzo (Instituto de Medicina Tropical, Universidad Central de Venezuela, Caracas, Venezuela) for synthesizing and donating the kemptide and kemptide-Lys8 peptides that were used in this work. We also thank Susan S. Taylor (Department of Chemistry, Biochemistry and Pharmacology, University of California San Diego, La Jolla, USA) for providing the SP20 and IP20 synthetic peptides that were employed here.
Financial support
This research was supported by grant numbers S1-IC-CB-007-14, S1-IC-CB-001-17 and S1-IC-CB-008-17 from Decanato de Investigación y Desarrollo, Universidad Simón Bolívar, Caracas, Venezuela, to JB, and 2013001659 from FONACIT, Caracas, Venezuela, to NAA.
Conflict of interest
None.
Ethical standards
We declare that all the experiments in this paper were carried out in accordance with the legal and ethical standards of Venezuela.