INTRODUCTION
Foodborne trematode infections in man occur worldwide, but are particularly common in certain regions due to dietary and cultural factors (Keiser and Utzinger, Reference Keiser and Utzinger2009). The Russian Far East is characterized by the presence of helminthiases not occurring elsewhere in the country; among the region-specific trematodes are digenean species of the genera Clonorchis and Metagonimus (Tatonova et al. Reference Tatonova, Chelomina and Besprosvannykh2012), Paragonimus and Nanophyetus (Besprozvannykh, Reference Besprozvannykh2002). Human nanophyetiasis is a zoonotic disease caused by members of the digenean genus Nanophyetus (Chapin, Reference Chapin1926), family Troglotrematidae Odhner, 1914. Their life cycles include a freshwater cerithioidean snail as the first intermediate host, a fish (usually a salmonid) as a second intermediate host and a variety of fish-eating animals as definitive hosts. The latter may include numerous species of carnivorous mammals, humans and, rarely, piscivorous birds (Millemann and Knapp, Reference Millemann and Knapp1970; Harrel and Deardorf, Reference Harrel and Deardorf1990; Coombs, Reference Coombs, Crompton and Savioli2006).
Nanophyetiasis has been reported on both Western and Eastern sides of the northern Pacific Rim, including the USA and Russian Federation (Ermolenko et al. Reference Ermolenko, Bespozvannykh, Rumyantseva and Voronok2015). Human infection with Nanophyetus spp. may cause abdominal discomfort, associated with nausea and vomiting, chronic diarrhoea, peripheral blood eosinophilia, weight loss and fatigue (Harrel and Deardorf, Reference Harrel and Deardorf1990). In North America, symptomatic human intestinal infections caused by Nanophyetus spp. were documented in Oregon, Washington and California among individuals who had ingested raw, undercooked or home-smoked salmon (Onchorhyncus species: e.g. Onchorhyncus tshawytscha, Onchorhyncus kisutch, Onchorhyncus mykiss, or their roe) (John and Petri, Reference John and Petri2006). In Russia, nanophyetiasis is commonly detected in the basins of the Amur River and Ussuri River and on Sakhalin Island (the Sea of Okhotsk). According to survey data, salmonid fishes are known to be the main food in the human population in areas with the highest incidence of disease. As a result, the infection among rural native populations (Evenki, Nivkhi) can reach 95–98% (Dragomeretskaia et al. Reference Dragomeretskaia, Zelia, Trotsenko and Ivanova2014). The prevalence of Nanophyetus infection may reach 82% among domestic carnivores and 17·6% among wild carnivores, with an intensity of infection reaching 19 000 digeneans per animal (Dragomeretskaia et al. Reference Dragomeretskaia, Zelia, Trotsenko and Ivanova2014). In North America, nanophyetiasis may be associated with a severe systemic illness in dogs called ‘salmon poisoning disease’ (SPD), which is usually fatal if left untreated. The cause of SPD is a rickettsial bacterium Neorickettsia helminthoeca, which uses Nanophyetus salmincola as the vector (Vaughan et al. Reference Vaughan, Tkach and Greiman2012). Recently, N. salmincola was demonstrated to carry at least two additional species of Neorickettsia pathogenic to carnivores (Greiman et al. Reference Greiman, Kent, Betts, Cochell, Sigler and Tkach2016). In contrast, no Neorickettsia symbionts have been reported from Eurasian Nanophyetus thus far.
Despite the broad distribution and medical and veterinary significance of these digeneans, the intrageneric taxonomy of Nanophyetus spp. remains unresolved. The genus includes 4 nominal species, N. salmincola (Chapin, Reference Chapin1926), Nanophyetus schikhobalowi Skrjabin and Podiapolskaia, 1931, Nanophyetus asadai (Yamaguti, 1971) and Nanophyetus japonensis Saito, Saito, Yamashita, Watanabe and Sekikawa, 1982. The latter two species were described from dogs only in Japan, are little known and apparently have very limited geographic distribution. Due to the morphological similarity of N. salmincola and N. schikhobalowi, their taxonomic status has always been unclear with different authors either considering them separate species or a single species distributed on two continents. At present, N. schikhobalowi is broadly assumed to be a synonym of N. salmincola (Bowman et al. Reference Bowman, Hendrix, Lindsay and Stephen2008). However, there has been no attempt to test this assumption using molecular tools.
In this study, we use sequence data and the secondary structure (SS) of the nuclear ribosomal RNA region to address the question of the taxonomic identity of the North American and Eurasian forms currently belonging to N. salmincola and their phylogenetic relationships.
MATERIALS AND METHODS
Parasite samples
Metacercariae were obtained from salmonid fishes (Brachymystax lenok Pallas, 1773) and minnows (Phoxinus oxycephalus Sauvage and Dabry de Thiersant, 1874), collected from Komissarovka and Ilistaya Rivers (Lake Khanka basin) and Komarovka River (Razdolnaya River basin) in the Russian Far East and from Coho salmon (Onchorhynchus kisutch Walbaum, 1792) collected from Willamette River, Oregon, northwestern USA (Fig. 1). Metacercariae from Far Eastern fishes were experimentally fed to rats, which were euthanized and examined 1 month after infection. Adult flukes were recovered from the small intestine, thoroughly rinsed in saline and fixed in 96% ethanol for molecular analysis and 70% ethanol for morphological studies. The specimens were identified based on their morphological characteristics.

Fig. 1. Specimen collecting areas in the Russian Far East and the Pacific Northwest of the USA.
DNA extraction, gene amplification and sequencing
Genomic DNA was extracted from 20 individual worms using the HotSHOT technique (Truett et al. Reference Truett, Heeger, Mynatt, Truett, Walker and Warman2000) for Russian samples and from 9 metacercariae using the ZR Genomic DNA tissue kit from Zymo Research (Irvine, California, USA) for American samples.
The complete 18S rDNA, internal transcribed spacers, ITS1-5·8S-ITS2 rDNA and partial 28S rDNA regions were amplified by polymerase chain reaction (PCR) using several sets of primers (Table 1) and either Taq polymerase from Sileks (Russia) or New England Biolabs (Ipswich, Massachusetts, USA) One-Taq Quick Load PCR mix.
Table 1. List of primers used for PCR and sequencing

The amplification of the complete 18S rDNA was done using primers 18S-E and 18S-F and annealing temperature of 58 °C; the amplification of the complete ITS region (ITS1-5·8S-ITS2 rDNA) was performed using primers 1/F and DigL1 and annealing temperature of 54 °C; the amplification of the complete 28S rDNA was performed using three pairs of external primers: DigL2 and 1500R, U1148 and L2450, U1846 and L3449 (Table 1) and annealing temperature of 55 °C in all cases. Amplified products were cleaned using either polyethylene glycol (PEG) 6000 solution (Harris, Reference Harris1992) or DNA Clean & Concentrator kit from Zymo Research.
PCR products (5 partial and 2 complete 18S rDNA, 24 complete ITS1-5·8S-ITS2 rDNA and 4 complete 28S rDNA) were sequenced directly using ABI BigDye chemistry (Applied Biosystems, Foster City, California, USA), alcohol-precipitated and run on an ABI Prism 3100 or 3130 automated capillary sequencers (Applied Biosystems). The PCR primers and additional internal primers (Table 1) were used for sequencing of corresponding PCR products (Fig. 2). Contiguous sequences were assembled using Sequencher (GeneCodes Corp., ver. 4·2), and submitted to GenBank under accession numbers LN871816-LN871817; LN852660-LN852663, LN871800-LN871815; LN871818 -LN871821 (Russian isolates; Table 2) and KX990278-KX990286 (Oregon isolates; Table 2).

Fig. 2. Positions of the primers used for PCR and sequencing in our study. See Table 1 for primer sequences.
Table 2. Sequences of rRNA genes and ITS (ITS1-5·8S-ITS2 rDNA) used in phylogenetic analyses
Statistical and phylogenetic analyses of genetic data
The newly-generated sequences and matching sequences of closely related taxa from other studies deposited in GenBank (Table 2) were aligned using the Clustal W program (Thompson et al. Reference Thompson, Higgins and Gibson1994). Genetic distances between species and phylogenetic relationships among taxa were obtained using Mega 5·1 (Tamura et al. Reference Tamura, Dudley, Nei and Kumar2007). Alignment gaps were included in the p-distance calculations. Phylogenetic inferences were also obtained through Bayesian Inference (BI) as implemented in MrBayes 3·1 (Ronquist and Huelsenbeck, Reference Ronquist and Huelsenbeck2003). Ambiguously aligned sites were excluded from the phylogenetic analyses. Modeltest 3·7 software (Posada and Crandall, Reference Posada and Crandall1998) was used to select the nucleotide substitution model for both Maximum likelihood (ML) and BI analyses. The TVM+G (Transversion Model plus Gamma) for ITS matrix, TrN+I for 18S and TPM3uf+G for 28S substitution models were used. Branch support values in ML were estimated by 1000 bootstrap replicates. In MrBayes, Markov Chain Monte Carlo (MCMC) chains were run for 10 million generations; 10 000 generations were discarded as burn-in. Obtained trees were rooted using complete ITS region (ITS1-5·8S-ITS2 rDNA), 18S rDNA and 28S rDNA sequences of Haplorchis pumilio (family Heterophyidae) Looss, 1896. Haplorchis pumilio was chosen as a member of the family that is basal, but not too distant, from the Troglotrematidae (Olson et al. Reference Olson, Cribb, Tkach, Bray and Littlewood2003).
SS prediction for aligned RNA sequences
The consensus structures for a set of aligned ITS1 and ITS2 sequences were computed using the program RNAalifold (Bernhart et al. Reference Bernhart, Hofacker, Ivo, Will, Gruber and Stadler2008). Additionally the folding of the ITS1 and ITS2 sequences into putative SS was performed with RNAfold (Hofacker et al. Reference Hofacker, Fontana, Stadler, Bonhoeffer, Tacker and Schuster1994) and Mfold version 3.0 (Zuker, Reference Zuker2003). Optimal consensus structures and base-pair probabilities were computed using the simple covariance scoring scheme which automatically, randomly samples mutations. In both cases we chose structures according to the universal principle of minimization of the free energy (MFE structures). RNA was folded at a fixed temperature of 37 °C, and the structure chosen from different output files had branching structures homologous to those previously published for other digenans (Morgan and Blair, Reference Morgan and Blair1995; Tatonova et al. Reference Tatonova, Chelomina and Besprosvannykh2012). Non-conserved base pairs and variable sites were detected within the structures. The individual SS of the ITS1 and ITS2 obtained by computing the thermodynamically most favourable folding in Mfold and RNAfold were consistent with their consensus predictions using RNAalifold.
RESULTS
Sequence analyses of 18S and 28S genes
The complete 18S rRNA gene of 2 specimens from the Russian population was 1893 bp long. Only partial 18S rDNA sequences (up to 1862 bp long) were obtained from 5 specimens from the American population. The aligned 18S rDNA sequences of the populations from the two continents differed from each other by only a single nucleotide, or 0·05%.
We sequenced the complete 28S rRNA gene of two specimens from the Russian population of Nanophyetus. The sequences were identical and 3872 bp long. The alignment of partial 28S rDNA sequences trimmed to the length of the shortest sequence was 1321 bp long and included 4 specimens from the Russian population and 1 specimen from the American population. The sequences from 2 continents differed in 7 positions or 0·5%.
Sequence analyses of ITS1, 5·8S rDNA and ITS2 sequences
The aligned ITS1-5·8S-ITS2 rDNA dataset included 20 complete sequences (1219 bp long) belonging to the Russian population and 5 complete sequences belonging to the American population (1186 bp long). The alignment was 1339 bp long and had several indel gaps. The sequences did not contain repeated motifs characteristic for the ITS1 region of some digenean groups (Van Herwerden et al. Reference Van Herwerden, Blair and Agatsuma1999; Tkach and Snyder, Reference Tkach and Snyder2008; Snyder and Tkach, Reference Snyder and Tkach2011). Specimens in the Russian population differed by 6 nucleotide substitutions (0·4%). The main type of substitutions was G↔C transversions, 3 of which were localized in the ITS1 and two were in 5·8S rDNA gene sequences. All sequences from the American population were identical. The divergence of ITS1-5·8S-ITS2 rDNA sequences between the Russian and American populations was much greater than that within the Russian population and reached 14% including 32 nucleotide substitutions and 139 bp from indels.
The ITS1 of Russian specimens was 772 bp long while the ITS1 of American specimens was 739 bp long. The variation in the ITS1 region between sequences of Nanophyetus spp. originating from different continents resulted from both nucleotide polymorphisms and size variation with ITS1 containing the majority of polymorphic positions. ITS1 sequences of the Russian and American populations differed by 27 nucleotide substitutions (4% of alignment length). The most common substitutions were transitions A↔G (37%) and T↔C (29%) while transversions C↔G and T↔A were much less common and constituted only 4% each; transversions A↔C and G↔T constituted 11% and 15% of all mutations, respectively. In the aligned ITS1 sequences we detected 15 indels: 10 short (1–6 bp), 3 medium-sized (8–19 bp) and 2 long (27 and 31 bp), totaling 139 bp. Thus, 17% of ITS1 alignment length consisted of indels (Supplementary Fig. S1). The genetic p-distance (d) estimated between the ITS1 sequences of the two populations, including alignment gaps was high (d = 0·2 or 20%).
ITS2 sequences in specimens from both Russian and American populations were 289 bp long. The variation of ITS2 sequences among the specimens was only due to nucleotide polymorphisms. There was only one nucleotide substitution (0·3% of sequence length) among Russian specimens while sequences of all specimens from the American population were identical. The 5 detected nucleotide substitutions among American and Russian populations were represented by 2 A↔G transitions, 2 T↔C transitions (40% each) and a single T→A transversion (20%). The genetic p-distance between Nanophyetus spp. populations in ITS2 was rather low at 0·02.
SS of the ITS intergenic spacers
The predicted SS for the complete ITS1 transcripts have a tree-like conformation and consist of two branches of complex structure that are separated by a helix (Figs 3, 4 and 5). The branch I, which is closer to 5′ end of ITS1 sequences, was more variable and contained the majority of nucleotide substitutions, while the branch II was more conserved and included trident-like helical structures in both Russian and North American digeneans. The most evident difference between the SS of ITS1 was the presence of 2 medium-sized hairpins in the helix connecting two branches in Nanophyetus from Russia (Fig. 3A). This pattern with some modifications was also observed in MFE models obtained from Mfold (Fig. 4). The RNAalifold free energy was lower than in Mfold for each SS reconstructed in our study. The calculated DG (the quantity of energy required for formation of the SS) in the two applications ranged from −276·3 to −264·2 in RNAalifold to −241·2 to −237·3 kcal mol−1 in Mfold (for Russian and American populations, respectively).

Fig. 3. De novo modelled predicted consensus secondary structures of ITS1 region reconstructed in RNAalifold: (A) Secondary structure for specimens from Russian population with a MFE of −276·30 (−276·30 plus 0·00 from covariance contributions) kcal mol−1; (B) Secondary structure for specimens from American population with a minimum free energy of −264·20 (−264·20 plus 0·00 from covariance contributions) kcal mol−1. Interspecific variable sites are in circles, intraspecific in squares, insertions are coloured grey, deletions marked with triangles. ITS, internal transcribed spacer; MFE, minimum free energy.
The ITS2 SS of the 24 Nanophyetus spp. modelled in both applications shared a model typical of digeneans, i.e. a palm with four fingers, first described in detail by Michot et al. (Reference Michot, Despres, Bonhomme and Bachellerie1993). Thus, the SS differed between the two compared populations (Fig. 5). While the folding patterns of A, B and D domains were the same, the longest, hairpin-shaped, domain C showed variation. Sequences from the Russian population had a large internal loop (marked with circle on Fig. 5A) near the apex of the domain C, while sequences from the American population had a similar loop at the top of the domain C (Fig. 5C). The DG required for formation of the SS in two applications ranged from −101·7 to −101·8 in RNAalifold to −94 to −94·5 kcal mol−1 in Mfold (for Russian and American populations, respectively).
Phylogenetic reconstructions
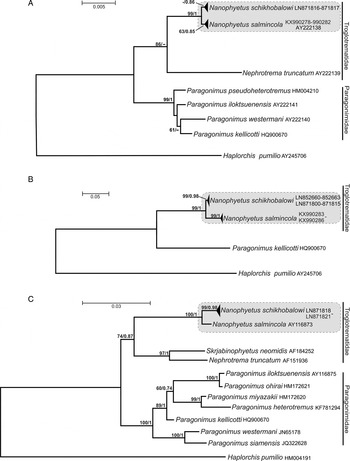
The phylogenetic tree based on the 18S rRNA sequence data clearly separated the Troglotrematidae and the Paragonimidae as sister taxa. Despite its conserved nature, this gene provided a strong support (99% bootstrap support in ML and 1·0 posterior probability in BI) for the Nanophyetus clade comprising both American and Russian specimens (Fig. 6A), although species-level clades received a somewhat lower support. The sequences of complete ITS1-5·8S-ITS2 rDNA region of the two forms of Nanophyetus obtained in this study fall into two strongly supported (99% bootstrap values in ML; 0·98–1·0 posterior probabilities in BI) monophyletic lineages corresponding to the two geographic areas of origins of the specimens (Fig. 6B). The Nanophyetus clade was also very strongly (99%/1) supported in this tree. Similarly, the 28S tree also clearly separated the Troglotrematidae and the Paragonimidae, but unlike the 18S tree, it provided a very strong support (99%/0·98) for the clade of Nanophyetus from Russia while the American Nanophyetus was represented in this dataset by a single sequence (Fig. 6C).
DISCUSSION
Nanophyetus has a complex taxonomic history that includes several revisions of the systematic position of the genus and its constituent species. Chapin (Reference Chapin1926) established the genus Nanophyes for the new species Nanophyes salmincola (Chapin, Reference Chapin1926) from dogs in Corvallis, Oregon (the area from which our American specimens were collected). In 1928 he changed the name to Nanophyetus salmincola because Nanophyes was preoccupied (Chapin, Reference Chapin1928). Chapin linked this new digenean species with the lethal ‘salmon poisoning disease’ of dogs endemic in the region. Later, this connection has been confirmed, although it was found that intracellular bacteria Neorickettsia helminthoeca symbiotic in the digeneans and not the digenean itself, were responsible for the disease (see Headley et al. Reference Headley, Scorpio, Vidotto and Dumler2011; Vaughan et al. Reference Vaughan, Tkach and Greiman2012 for reviews). At the time of description the genus was included in the family Heterophyidae Odhner, 1914. A metacercaria named Distomulum oregonensis was described by Ward and Mueller in 1926, slightly later than the Chapin's (Reference Chapin1926) new species, and thus synonymized with Nanophyetus salmincola. Several years later, a morphologically similar species Nanophyetus schikhobalowi Skrjabin and Podiapolskaia, 1931 was described in the Far East of the former Soviet Union, now the Russian Federation (Sinovich, Reference Sinovich1959). Although N. schikhobalowi seemed to differ from N. salmincola in several morphological characteristics, primarily in having smaller eggs, Gebhardt (Reference Gebhardt1966) concluded that these two species were synonymous. This view was accepted by a number of researchers, some of whom agreed on assigning the Eurasian form a subspecies status as N. salmincola schikhobalowi (Filimonova, Reference Filimonova1966; Gebhardt, Reference Gebhardt1966). Additionally, some authors considered the genus Nanophyetus a synonym of Troglotrema Odhner, 1914 (Kinne, Reference Kinne1980; Bowman et al. Reference Bowman, Hendrix, Lindsay and Stephen2008). Hence, the question of the content of Nanophyetus and taxonomy of the nominal species within the genus remained unresolved due to ambiguous morphological differences.
In the present study, we compared Nanophyetus spp. from the Russian Far East and North America using multi-locus molecular data. The ITS1 and ITS2 of the nuclear ribosomal DNA region are among the most frequently used markers in differentiation among digenean species (Nolan and Cribb, Reference Nolan and Cribb2005; Razo-Mendivil et al. Reference Razo-Mendivil, Vázquez-Domínguez, Rosas-Valdez, de León and Nadler2010), therefore our differentiation was primarily based on this region. The levels of sequence divergence between these populations correspond to the levels of interspecific differences observed in various digenean taxa (Morgan and Blair, Reference Morgan and Blair1995; Tkach and Snyder, Reference Tkach and Snyder2007, Reference Tkach and Snyder2008, Reference Snyder and Tkach2011; Brant and Loker, Reference Brant and Loker2009; Curran et al. Reference Curran, Tkach and Overstreet2013; Tkach et al. Reference Tkach, Curran, Bell and Overstreet2013; Kasl et al. Reference Kasl, Fayton, Font and Criscione2014). Variability observed among individuals of the Russian form in our study corresponded well to the intraspecific variability previously observed within digenean species. This variability usually does not exceed 1% in the ITS1-5·8S-ITS2 rDNA region, although higher levels of variability have also been reported (Nolan and Cribb, Reference Nolan and Cribb2005; Tkach et al. Reference Tkach, Curran, Bell and Overstreet2013; Kasl et al. Reference Kasl, Fayton, Font and Criscione2014).
The level of differences between Russian and American specimens of Nanophyetus was much higher than intraspecific variability and reached 14% in the ITS1-5·8S-ITS2 rDNA region which indicates that the two forms definitely belong to different species. For example, published interspecific differences in ITS1-5·8S-ITS2 rDNA region ranged from 0·5–7·8% among congeneric digeneans belonging to various digenean genera, e. g., Trichobilharzia, Schistosoma (family Schistosomatidae) Diplostomum (Diplostomatidae), Haematoloechus (Haematoloechidae), Homalometron (Apocreadiidae), Echinostoma (Echinostomatidae), Macroderoides (Macroderoididae), Aptorchis (Plagiorchiidae) and Alloglossidium (Alloglossidiidae) (Morgan and Blair, Reference Morgan and Blair1995; Tkach and Snyder, Reference Tkach and Snyder2007, Reference Tkach and Snyder2008; Brant and Loker, Reference Brant and Loker2009; Curran et al. Reference Curran, Tkach and Overstreet2013; Tkach et al. Reference Tkach, Curran, Bell and Overstreet2013; Kasl et al. Reference Kasl, Fayton, Font and Criscione2014). Van Herwerden et al. (Reference Van Herwerden, Blair and Agatsuma1999) have demonstrated rather high interspecies variation in the ITS1 region of Paragonimus, a genus closely related to Nanophyetus. To the best of our knowledge, there have been no newer data since then. Therefore, based on the combination of the newly obtained molecular evidence and previously published morphological differences, we restore the validity of N. schikhobalowi as an independent species.
The interspecific variation in ITS1 and ITS2 rDNA sequences in our study reached 20 and 2%, respectively. For comparison, the interspecific sequence divergence of the ITS1 spacer among members of the Mesometridae ranged from 6·6 to 19·1%, which was only slightly higher than that of the ITS2 of the same digenean species, which reached 3·4–15·1% (Jousson et al. Reference Jousson, Bartoli, Zaninetti and Pawlowski1998). Nolan and Cribb (Reference Nolan and Cribb2005) noticed that levels of the interspecific variation of the whole ITS region is typically substantial. However, a number of studies have found very low differences between some congeneric species, especially in the ITS2. For example, the ITS2 sequences reported for species of Schistosoma and Fasciola differed by only 0·3–1·7% within each genus (Lotfy et al. Reference Lotfy, Brant, DeJong, Le, Demiaszkiewicz, Rajapakse, Mareka, Zouhara, Doudab, Mazakovaa and Rysanek2010). High interspecific divergence in the ITS1 region of Nanophyetus resulted from both nucleotide substitutions and indels. Significant interspecific differences in the length of ITS1 were reported for species of Dolichosaccus (family Plagiorchiidae), Schistosoma (Schistosomatidae), Aptorchis (Plagiorchiidae), Choanocotyle (Choanocotylidae) and other digeneans (Tkach and Snyder, Reference Tkach and Snyder2007 and references therein).
SS of ITS1 and ITS2 is important for the correct processing of mature rRNA and biogenesis of active ribosomal subunits (Michot et al. Reference Michot, Despres, Bonhomme and Bachellerie1993; Hofacker et al. Reference Hofacker, Fontana, Stadler, Bonhoeffer, Tacker and Schuster1994; Koetschan et al. Reference Koetschan, Förster, Keller, Schleicher, Ruderisch, Schwarz, Müller, Wolf and Schultz2010; Ghatani et al. Reference Ghatani, Shylla, Tandon, Chatterjee and Roy2012). Therefore, it is a useful additional source of biological information assisting in differentiation between taxa. In our assessment of the rRNA SS in Nanophyetus spp. we used the optimal structures generated by Mfold and RNAfold software while paying attention to the similarity of folding conditions. RNA was folded at a fixed temperature of 37 °C and the structures with the highest negative free energy were chosen from output files. As expected in closely related taxa, the SS of ITS1 and ITS2 regions revealed in the present study had common features in the two Nanophyetus species. However, they also had unique characteristics, such as the presence of 2 hairpins in the central helix of ITS1 in worms from the Russian population (Fig. 3) as well as differences in the number, shape and size of the loops that connect stems. Differences in the topology are mainly due to differences in sequence lengths resulting from the large number of indels. Unfortunately, comparative studies of SS of the ITS region in parasitic flatworms are scarce (Capriotti and Marti-Renom, Reference Capriotti and Marti-Renom2008; Lotfy et al. Reference Lotfy, Brant, DeJong, Le, Demiaszkiewicz, Rajapakse, Mareka, Zouhara, Doudab, Mazakovaa and Rysanek2010; Tatonova et al. Reference Tatonova, Chelomina and Besprosvannykh2012). The latter authors described SS of the ITS1 transcripts of Clonorchis sinensis as branching stems resembling those observed in the present work.
Due to the presence of several conserved sequence regions and structural elements within ITS2, a number of taxa share a similar pattern of ITS2 folding. SS of ITS2 in Nanophyetus showed the typical four-domain model reported for a number of different organisms including trematodes (Michot et al. Reference Michot, Despres, Bonhomme and Bachellerie1993; Morgan and Blair, Reference Morgan and Blair1995; Tatonova et al. Reference Tatonova, Chelomina and Besprosvannykh2012). SS of ITS1 and ITS2 revealed in the present study had some shared features as well as unique characteristics that clearly distinguished species from Russian and American populations (Figs 3–5).

Fig. 4. De novo modelled individual predicted MFE secondary structures of ITS1 region reconstructed in Mfold: (A) Secondary structure for specimens from Russian population with a MFE of −241·20 kcal mol−1; (B) Secondary structure for specimens from American population with a MFE of −237·30 kcal mol−1. Interspecific variable sites are in circles, intraspecific in squares, insertions are coloured grey. MFE, minimum free energy.
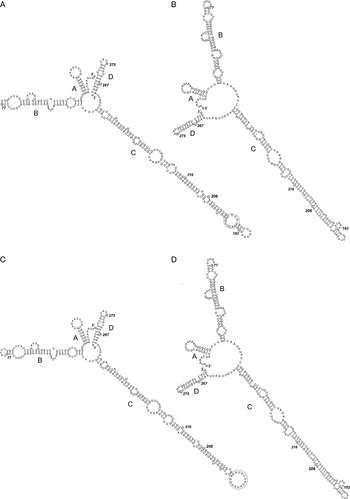
Fig. 5. De novo modelled predicted secondary structures of ITS2 region: (A) Consensus secondary structure with a MFE of −101·70 (−102·10 plus 0·40 from covariance contributions) kcal mol−1 for specimens from Russian population reconstructed in RNAalifold; (B) MFE secondary structure with a MFE of −94 kcal mol−1 for specimens from Russian population reconstructed in Mfold; (C) Consensus secondary structure with a MFE of −101·80 (−101·80 plus 0·00 from covariance contributions) kcal mol−1 for specimens from American population reconstructed in RNAalifold; (D) MFE secondary structure with a MFE of −95·4 kcal mol−1 for specimens from American population reconstructed in Mfold. Interspecific variable sites are in circles; intraspecific in squares; invariable loops are in black circles. MFE, minimum free energy.
Phylogenetic trees based on different regions of nuclear rDNA used in our analyses produced identical tree topologies. However, the level of branch support was different depending on the DNA region used. The conserved 18S gene differentiated well between Nanophyetus and other genera, but did not provide a significant separation between the two Nanophyetus species (Fig. 6A). The ITS1-5·8S-ITS2 rDNA region has provided a high level of support for the clades representing N. salmincola and N. schikhobalovi. The same is true for the N. schikhobalovi clade (99/0·98 support) in the 28S tree, but N. salmincola in this tree was represented by only one sequence. This corroborates the high utility of ITS markers for species differentiation and phylogenetic analyses at intrageneric and intra-familial levels in this digenean lineage.

Fig. 6. Maximum Likelihood phylogeny of Nanophyetus based on (A) 18S rDNA sequences; (B) ITS1+5·8S+ITS2 rDNA sequences; (C) 28S rDNA sequences. Statistical branch support values are as follows: ML bootstrap values/BI posterior probabilities. Only bootstrap support values greater than 50% in ML and posterior probabilities greater than 0·7 in BI are shown. ITS, internal transcribed spacer; ML/BI, Maximum likelihood/Bayesian inference.
Concluding remarks
The present study used multi-locus DNA sequence data (18S, ITS1+5·8S+ITS2 and 28S rDNA) and the genetic diversity of the SS of the ITS spacers to analyze interrelationships and taxonomic status of North American and Eurasian forms of Nanophyetus previously considered different sub-species of the same broadly distributed species. The data obtained have provided strong evidence for the status of these forms as independent species, N. salmincola and N. schikhobalowi, with disjunct distribution ranges. These results may have implications for understanding of their host associations and pathogenicity including the ability to carry agents of ‘salmon poisoning disease’ of dogs and other Neorickettsia that are known from Nanophyetus salmincola, but not from N. schikhobalowi. These differences are intriguing and require future in-depth studies.
SUPPLEMENTARY MATERIAL
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182016002171.
ACKNOWLEDGEMENTS
The authors are grateful to Dr Michael Kent (Oregon State University, Corvallis, USA) for providing samples of Coho salmon kidneys infected with Nanophyetus metacercariae and Dr Stephen Greiman (University of North Dakota) for his assistance with DNA extraction from these samples. We thank Dr John M. Kinsella for his comments and English corrections in the manuscript.
FINANCIAL SUPPORT
This work was supported in part by the grant R15AI092622 from the National Institutes of Health, USA to VVT and by the FEB RAS (Far Eastern Branch Russian Academy of Sciences) project number 15-I-6-014.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICAL STANDARDS
Animal use procedures were approved by the Institutional Committee for Laboratory Animal Use and Care of the Institute of Biology and Soil Science, Far Eastern Branch, Russian Academy of Sciences.