Introduction
Establishing species and lineage boundaries by phylogenetics and population genetics is a central task in evolutionary biology and has become mandatory when studying emerging pathogens and neglected infectious diseases (Yang and Rannala, Reference Yang and Rannala2010). How parasite lineages and species are related to each other in space and time can provide valuable insights, such as pathogen adaptation to their hosts, the role of genetic exchange and/or clonality in emergence of novel genotypes, and spatial distribution of genetic variation; these features are often related to morbidity, transmissibility, or drug resistance (De Meeûs et al., Reference De Meeûs, McCoy, Prugnolle, Chevillon, Durand, Hurtrez-Bousses and Renaud2007).
Chagas disease (CD) is a complex zoonosis caused by the protozoan parasite Trypanosoma cruzi and infects nearly 8 million people in Latin America, with another 25 million people currently at risk of acquiring the infection (WHO, 2016). According to World Health Organization, CD remains the largest parasitic disease burden in the Western Hemisphere (WHO, 2016). In addition, natural T. cruzi transmission cycles are complex, with notable diversity of triatomine vectors and mammalian hosts of virtually all orders that interact between sylvatic and domestic cycles (Jansen et al., Reference Jansen, Xavier and Roque2017; Justi and Galvão, Reference Justi and Galvão2017).
T. cruzi is a successful parasite that displays extraordinary intraspecific genetic diversity, with at least six lineages, or discrete typing units (DTUs TcI–TcVI), currently recognized and distributed throughout the Americas (Zingales et al., Reference Zingales, Miles, Campbell, Tibayrenc, Macedo, Teixeira and Sturm2012), in addition an emergent and well-supported DTU closely related to TcI that is mostly found in bats (TcBat) (Marcili et al., Reference Marcili, Lima, Cavazzana, Junqueira, Veludo, Maia Da Silva and Teixeira2009; Lima et al., Reference Lima, Espinosa-Álvarez, Ortiz, Trejo-Varón, Carranza, Pinto, Serrano, Buck, Camargo and Teixeira2015; Ramírez et al., Reference Ramírez, Hernández, Montilla, Zambrano, Flórez, Parra and Cucunubá2014). Among these DTUs, TcV and TcVI are considered hybrids of parental groups TcII and TcIII, and are linked to domestic cycles and human infections in southern cone countries (Brisse et al., Reference Brisse, Henriksson, Barnabé, Douzery, Berkvens, Serrano and Tibayrenc2003; Lewis et al., Reference Lewis, Llewellyn, Yeo, Acosta, Gaunt and Miles2011). Moreover, it was proposed TcI and TcII are the natural ancestors of the species and ancient recombination events that produced TcIII and TcIV (Westenberger et al., Reference Westenberger, Barnabé, Campbell and Sturm2005).
TcI is the most geographically widespread and diverse lineage, with overlapping distributions between domestic and sylvatic cycles (León, Reference León, Hernández, Montilla and Ramírez2015; Ramírez and Hernández, Reference Ramírez and Hernández2018). Recently, based on multiloci approaches, a clearly divergent and homogeneous TcI genotype associated with human infections (TcIDOM) was described (Llewellyn et al., Reference Llewellyn, Miles, Carrasco, L, Yeo, Vargas and Gaunt2009c; Ramírez et al., Reference Ramírez, Guhl, Messenger, Lewis, Montilla, Cucunuba and Llewellyn2012; Ramírez et al., Reference Ramírez, Tapia-Calle and Guhl2013). Bayesian skyline plots proposed TcIDOM divergence and expansion ~23–12 KYA, which is consistent with the first human settlements in America (Ramírez et al., Reference Ramírez, Guhl, Messenger, Lewis, Montilla, Cucunuba and Llewellyn2012). It was also suggested that TcIDOM first made contact with humans in North–Central America and subsequently become widespread throughout South America (Zumaya-Estrada et al., Reference Zumaya-Estrada, Messenger, Lopez-Ordonez, Lewis, Flores-Lopez, Martínez-Ibarra and Llewellyn2012).
Nevertheless, despite intensive efforts to elucidate the remarkable genetic diversity of T. cruzi DTUs, there are some gaps related to the understanding of the origin, relationships, and ecological and epidemiological relevance of the DTUs [reviewed in (Messenger et al., Reference Messenger, Llewellyn, Bhattacharyya, Franzén, Lewis, Ramírez and Miles2015; Brenière et al., Reference Brenière, Waleckx and Barnabé2016)]. There are a few reasons for these knowledge gaps. First, there is scarce information about the parasite's geographical distribution and ecotopes, including the vast variety of sylvatic hosts that remain unsampled, especially at local and microgeographic scales. Second, use of different markers (nuclear and mitochondrial) has led to different genealogical histories and thus to distinct inter- and intra-DTU division proposals, ranging among geography, transmission cycles, or vertebrate/invertebrate hosts [e.g. (Herrera et al., Reference Herrera, Bargues, Fajardo, Montilla, Triana, Vallejo and Guhl2007, Reference Herrera, Guhl, Falla, Fajardo, Montilla, Adolfo Vallejo and Bargues2009; Llewellyn et al., Reference Llewellyn, Miles, Carrasco, L, Yeo, Vargas and Gaunt2009c; Ramírez et al., Reference Ramírez, Tapia-Calle and Guhl2013)]. In addition, it is widely recognized that the majority of gene trees may be incongruent with the true underlying species tree, real speciation events, and within species population structure (Fujita et al., Reference Fujita, Leaché, Burbrink, McGuire and Moritz2012; Leaché et al., Reference Leaché, Koo, Spencer, Papenfuss, Fisher and McGuire2009). Third, despite the current technological advances for collecting genomic datasets that include high-resolution markers and approaches [e.g. nuclear Multilocus Sequence Typing (MLST), mitochondrial MLST, and Multilocus Microsatellite Typing (MLMT)], phylogenetic methods to describe T. cruzi diversity and DTU relationships are mainly based on clustering algorithms and/or concatenation [e.g. (Llewellyn et al., Reference Llewellyn, Lewis, Acosta, Yeo, Carrasco, Segovia and Gaunt2009a; Flores-López and Machado, Reference Flores-Lopez and Machado2011; Yeo et al., Reference Yeo, Mauricio, Messenger, Lewis, Llewellyn, Acosta and Miles2011; Diosque et al., Reference Diosque, Tomasini, Lauthier, Messenger, Monje Rumi, Ragone and Yeo2014)]. Although concatenation is a heuristic strategy that provides phylogenies with high resolution, it assumes that the evolutionary history of each gene tree is identical to the species tree (Ogilvie et al., Reference Ogilvie, Bouckaert and Drummond2017). While it is very true that incongruence is frequently observed between individual loci in T. cruzi, most recent publications using MLST, first utilize software (e.g. MLSTest), to detect statistically significant incongruence. Concatenation decisions are then based upon the absence of significant evidence for independent evolutionary histories of particular loci. This fact must be acknowledged, that this is one way to circumvent these limitations. However, novel methodologies should be explored in order to fill the gaps and underpin a better understanding of the evolutionary history of the DTUs and in particular TcI.
To help fill some of the knowledge gaps described above and avoid gene tree/species conflicts, we implemented a multispecies coalescent (MSC) approach to infer intra-TcI relationships and determine lineage boundaries testing the phylogenetic position of TcIDOM, and included previously reported isolates and new isolates from Mexico, Brazil, and Argentina. We conducted the first evaluation (Using MSC) of the current position of TcIDOM as an independent genotype of the TcI complex. Moreover, using a set of different independent loci, including housekeeping genes, we assessed for possible incongruence in gene genealogies and examined intra-lineage diversity through genetic diversity and network analyses.
Materials and methods
Trypanosome culture, sequencing, and molecular data collection
T. cruzi isolates (Tables 1 and 2) were cryopreserved in liquid nitrogen and maintained in LIT medium, pH 7.4, and supplemented with 10% fetal bovine serum (v/v) for DNA preparation. Genomic DNA of cultured trypanosomes was extracted from pellets of approximately 106 parasites using the GeneJet kit (Thermo Scientific®), according to the manufacturer's instructions. DNA amplification of the desired region was done in a thermocycler (PTC-100 MJ Research®) using a 50-μL PCR mix as follows: 100 ng of DNA, 1 U of Phusion High-Fidelity DNA Polymerase (Thermo Scientific®) with 5 µL of Phusion HF Buffer, and 1 mm of each dNTP. PCR conditions were subjected to an initial denaturation temperature of 95 °C/10 min followed by 40 cycles (denaturation at 95 °C for 1 min; annealing temperature for 1 min as indicated in Supplementary file 1; elongation at 72 °C for 1 min). Amplicons were precipitated in 70% ethanol and suspended in water. Sequencing was done by the dideoxy-terminal method in an automatic sequencer (AB3730, Applied Biosystems® Genetic Analyzer) by both strands at ACTGene Molecular Analysis (Brazil).
Table 1. Cytb sequences used in this study

Table 2. SL-IR sequences and isolates used in this study

Resulting sequences with expected sizes of each marker (Table 3) were employed to generate multiple sequence alignments using Muscle v.3.8 (Edgar, Reference Edgar2004) with default settings, and manually edited in GeneDoc v.2.6.01 (Nicholas et al., Reference Nicholas, Nicholas and Deerfield1997). Based on previously reported TcI sequences from GenBank, we built different sets of alignments for phylogenetic and population genetic analyses. GenBank accession numbers, hosts, and geographical origin for the samples are listed in Tables 1 and 2 and include the new sequences derived from this study.
Table 3. Molecular data obtained in this study

a Nucleotide substitutions models were selected under a Bayesian framework in bModelTest (see (47) for details).
Isolates and sequences included in the study
This study mainly examined TcI complex genetic diversity, relationships based on previously reported sequences in GenBank, and by applying recent advances in Bayesian phylogenetic species tree estimation (MSC), we also examined genetic variation patterns in 20 new isolates from Mexico [9], Brazil [5], and Argentina [6] (Tables 1 and 2). We sequenced three nuclear regions [TcSC5D, a putative lathosterol/episterol oxidase (Cosentino and Agüero, Reference Cosentino and Agüero2012); TcMK, mevalonate kinase (Cosentino and Agüero, Reference Cosentino and Agüero2012); and SL-IR, spliced leader intergenic region of the miniexon gene (Burgos et al., Reference Burgos, Altcheh, Bisio, Duffy, Valadares, Seidenstein and Schijman2007)] and two mitochondrial genes [Cytb, cytochrome b (Messenger et al., Reference Messenger, Llewellyn, Bhattacharyya, Franzén, Lewis, Ramírez and Miles2012); NTR, Nitroreductase (Hall et al., Reference Hall, Meredith and Wilkinson2012)]. We selected these housekeeping genes because of their lack of availability in parasite databases and their importance in the maintenance of parasite cellular functions.
Because of the lack of availability of TcMK, TcSC5D, and NTR genes in GenBank, only parasite isolates from our work were considered when evaluating these genes, except for those from Brazil. MS, SP: Brazilian States, Mato Grosso do Sul and São Paulo, respectively.
Parasite genetic diversity and network analysis
We phased TcMK, NTR, Cytb, TcSC5D, and SL-IR loci in DNAsp v.5.10.01 (Librado and Rozas, Reference Librado and Rozas2009) with a threshold of 0.9 and 300 iterations, although several exploratory analyses yielded identical results. General sequence diversity statistics for each locus were calculated in DNAsp v.5.10.01, including number of haplotypes, h; haplotype diversity, Hd; number of polymorphic sites, S; and nucleotide diversity, π.
To infer possible associations within and between TcI isolates from our data and those previously reported in GenBank (Tables 1 and 2; Fig. 1), resulting haplotypes of Cytb and SL-IR were employed to construct haplotype networks by the median-joining network method in popART v.1.7 (Leigh and Bryant, Reference Leigh and Bryant2015) and were selected if ε values ranged from 0.5 to 1. For both analyses, sites with gaps were excluded, including the microsatellite motif in SL-IR, as previously suggested (Tomasini et al., Reference Tomasini, Lauthier, Rumi, Ragone, D'Amato, Brandan and Diosque2011).

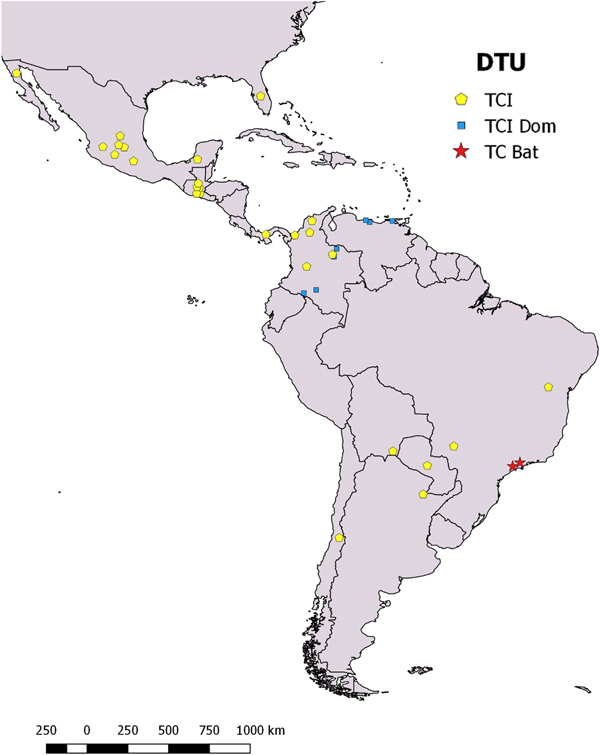
Fig. 1. Geographical origin of TcI isolates employed in this study across the endemic distribution of Chagas disease in the Americas. TcI, TcIDOM, and TcBat are indicated by color. The coordinates of each isolates were used to build a georeferenced map of isolates location. The map was built on ArcGIS10.3 using Esri Colombia PublicadorSIG layer (http://www.arcgis.com/home/item.html?id=b051fbef7fba406fbb8e62b90925f365#overview).
Intra-lineage relationships: gene and species tree estimation
The main goal of coalescent-based methods is to identify independently evolving lineages (Fujita et al., Reference Fujita, Leaché, Burbrink, McGuire and Moritz2012). Unlike concatenated phylogenies, which assume that gene trees match for all loci in species trees (a process often called reciprocal monophyly), the MSC approach uses multilocus sequence data in a Bayesian framework and accounts for gene tree discordance by modeling coalescent stochasticity in considered populations (Yang and Rannala, Reference Yang and Rannala2010). Consequently, evolutionary lineages can be identified and species trees can be precisely estimated, even in the absence of monophyly or when the phylogenetic signal present in the loci is weak, especially because of recent divergence processes (Degnan and Rosenberg, Reference Degnan and Rosenberg2009).
Thus, we estimated gene and species tree topologies using a Bayesian MSC approach using StarBEAST2 (Ogilvie et al., Reference Ogilvie, Bouckaert and Drummond2017) and implemented in BEAST v.2.4.7 (Bouckaert et al., Reference Bouckaert, Heled, Kühnert, Vaughan, Wu, Xie and Drummond2014). This multilocus method co-estimates the gene trees embedded in a shared species tree and does not require that each gene alignment/sample has the same number of sequences, and only requires that each sequence in each gene alignment is mapped to the appropriate species (Heled and Drummond, Reference Heled and Drummond2009; Ogilvie et al., Reference Ogilvie, Bouckaert and Drummond2017). StarBEAST2 enables faster species tree inference and more accurate estimates of substitution rates compared with previous versions of StarBEAST and likelihood-based methods that include concatenation (Ogilvie et al., Reference Ogilvie, Bouckaert and Drummond2017). Phylogenetic comparative methods as MSC that incorporate intraspecific variability are relatively new and, so far, not especially widely used in empirical studies. MSC is also helpful in depicting and explaining the genetic diversity signals and useful to infer intraspecific relationships.
For the analysis, TcI sequences of each gene alignment were previously mapped as TcI, TcBat, or TcIDOM based on genotyping assignment and earlier studies (Tables 1 and 2). Trypanosoma cruzi marinkellei and T. dionisii were used as outgroups. Models of DNA evolution were determined using bModelTest (Bouckaert and Drummond, Reference Bouckaert and Drummond2017) (Table 3) using the transition/transversion split setting. We used BModelAnalyser (Bouckaert et al., Reference Bouckaert, Heled, Kühnert, Vaughan, Wu, Xie and Drummond2014) to visualize bModelTest log output, after discarding 20% as burn-in. Because divergence time estimates were not goals of this study, we used an uninformative strict clock prior (1/×) for each gene to simplify the model and help the analysis converge. Additionally, we ran two independent MCMC runs of 2.2 × 107 generations each, with a sample frequency of 1000 and using a Yule tree prior with constant population sizes. Convergence of the chains were checked in Tracer v.1.6 (Rambaut et al., Reference Rambaut, Drummond, Xie, Baele and Suchard2018) (effective sample sizes >200). The two replicated analyses were combined in LogCombiner v.2.2 (Bouckaert et al., Reference Bouckaert, Heled, Kühnert, Vaughan, Wu, Xie and Drummond2014) after discarding the first 10% of trees as burn-in. Final trees were summarized in a maximum clade credibility tree using TreeAnnotator v.2.4.7 (Bouckaert et al., Reference Bouckaert, Heled, Kühnert, Vaughan, Wu, Xie and Drummond2014) and edited in iTol v.3 (Letunic and Bork, Reference Letunic and Bork2016).
Results
Molecular data collection and parasite isolates
In this study, we sequenced 20 parasitic isolates for five molecular markers, all of which were previously genotyped as a TcI DTU (Gómez-Hernández et al., Reference Gómez-Hernández, Rezende-Oliveira, Nascentes, Batista, Kappel, Martinez-Ibarra and Ramírez2011; Lauthier et al., Reference Lauthier, Tomasini, Barnabé, Rumi, D'Amato, Ragone and Diosque2012) (Tables 1 and 2). The resulting sequences were deposited in Genbank under the accession numbers (MH549646-MH549700). The five loci employed varied in length from 262 to 734 bp, and represented a total of 2917 bp, and included 264 and 206 variable and parsimony-informative sites, respectively (Table 3). It is worth noting that it was not possible to amplify Brazilian isolates for genes other than SL-IR and Cytb because some loci were recalcitrant to PCR amplification, possibly because of mutations in some regions of the primers (Supplementary file 1). Moreover, we found evidence of an indel (12-bp long) in the NTR gene from the Mexican INC-5 isolate (Supplementary file 2).
Parasite genetic diversity and network analysis
Overall, based on our dataset, we found evidence of high genetic diversity among TcI strains isolated from Mexico, Brazil, and Argentina (Table 4), but relatively low π estimates in contrast with high Hd indicates a certain amount of similarity between considered strains. Interestingly, Mexican isolates sampled for TcSC5D showed a unique haplotype and no evidence of polymorphic sites (Table 4). Despite the results obtained, genetic diversity estimates were not delimited by country or geography because each gene yielded different estimates [Cytb: Argentina < Brazil < Mexico; SL-IR: Mexico < Argentina < Brazil; TcSC5D: Mexico < Argentina; TcMK: Argentina < Mexico; NTR: Argentina < Mexico].
Table 4. Genetic diversity measures delimited by country

n: number of sequences analyzed; S: number of polymorphic sites; h: number of haplotypes; Hd: haplotype diversity; π: nucleotide diversity; s.d.: standard deviation.
To infer associations between shared Cytb and SL-IR alleles and haplotypes, we included a representative set of sequences from North–Central America, Northern South America, and Southern South America that belong to the endemic CD distribution (Tables 1 and 2; Fig. 1). Cytb and SL-IR haplotype networks showed no evidence of genetic structure based on geography or host (Fig. 2A and B). Thus, our Cytb haplotypes shared alleles between Mexico, Guatemala, Panama, Colombia, Venezuela, Brazil, and Argentina (H5; Supplementary file 3) and other related haplotypes with short mutational steps (Fig. 2A). However, other haplotypes from our Mexican isolates (KR1, CHG1, CGH2, CGH3, and CGH4) and Brazilian isolates AQ1–7 were strongly isolated and separated by several mutational steps (Fig. 2A). Moreover, Cytb had quite homogeneous and divergent haplotypes between TcIDOM and TcBat (Fig. 2A). Similarly, the SL-IR haplotype network (Fig. 2B) shared mixed haplotypes between Mexico, Colombia, the United States, and Argentina, and grouped closed to Paraguayan haplotypes (H8–H9; Supplementary file 4; Bolivia and Argentina had mixed haplotypes (H34; Table 2; Supplementary file 4).

Fig. 2. haplotypes inferred by the median-joining network method in popART v.1.7 (A). Cytb network; (B). SL-IR network. Size of the circles corresponds to the frequency of isolates per haplotype, and length of the vertical lines that connect the networks represents the number of mutations. Black dots indicate the median vectors, which include inferred ancestral nodes.
Gene trees and species tree discordance: phylogenetic position of TcIDOM
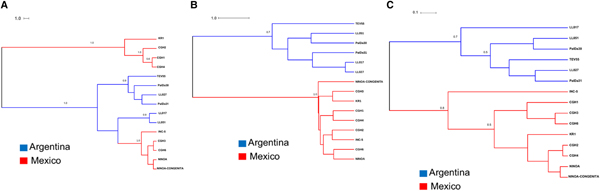
Our results support high phylogenetic incongruence between gene trees and embedded species tree topologies (Figs 3–6). Additionally, we found evidence of incongruence between our parasite isolates and gene trees (Figs 3–5). Consequently, the TcMK gene tree (Fig. 3A) showed also no strict association with either geography or host, and isolates were grouped into two clades: the first monophyletic clade included isolates KR1, CGH2, CGH1, and CGH4 from Mexico, and the second clade included Argentinean and Mexican isolates (CGH3, NINOA, and NINOA CONGENITA) (Fig. 3A). In contrast, TcSC5D and NTR gene trees showed two clades that were clearly separated by country, but with different tree topologies and branch lengths (Fig 3B and C).

Fig. 3. Maximum Clade Credibility (MCC) Tree of TcI isolates inferred by StarBeast2. (A). Tree constructed using the TcMK (Mevalonate Kinase) sequences; (B). Tree constructed using the TcSC5D (putative lathosterol/episterol oxidase) sequences; (C). Tree constructed using the NTR (Nitroreductase) sequences.

Fig. 4. Cytb Maximum Clade Credibility (MCC) Tree of TcI isolates inferred by StarBeast2. Numeric values correspond to posterior probability ranging from 0 to 1.

Fig. 5. SL- IR Maximum Clade Credibility (MCC) Tree of TcI isolates inferred by StarBeast2. Numeric values correspond to posterior probability ranging from 0 to 1.

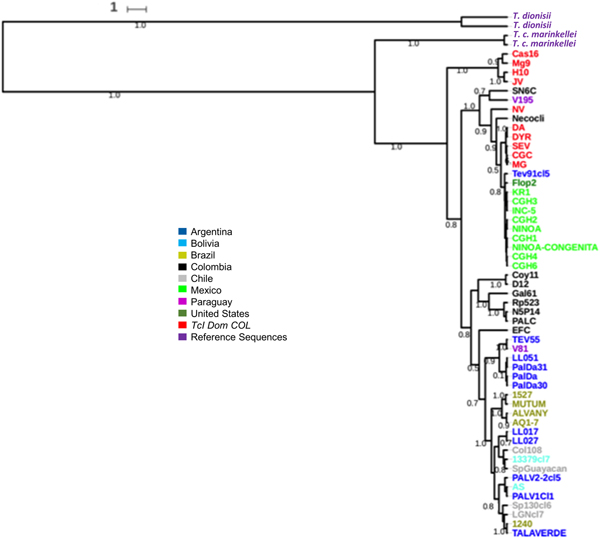
Fig. 6. Maximum Clade Credibility (MCC) species tree recovered from StarBeast2 analysis, illustrating Intra-TcI relationships and using T. dionisii and T. c. marinkellei as outgroups. Numeric values correspond to posterior probability ranging from 0 to 1.
Based on both the SL-IR and Cytb gene trees (Tables 1 and 2), TcIDOM from Colombia and Venezuela were not monophyletic (Figs 4 and 5). However, Cytb gene clearly differentiated TcIDOM Colombia from other TcI isolates, whereas TcIDOM Venezuela clustered with Mexican and Central American isolates (Fig. 4). Something similar was observed for SL-IR and TcIDOM Colombia (Fig. 5). Regardless, the gene trees did not reject TcIDOM as a discrete and emergent TcI genotype. The species tree recovered strong posterior probability values for TcI and TcIDOM (Fig. 6).
Discussion
A comprehensive understanding of T. cruzi epidemiology across its geographical range based on genetic diversity and phylogeny is essential for elucidating the parasite's evolution and natural history, which in turn provides important insights for further diagnosis, treatment, prevention and control efforts. Our results here support the emergence of TcIDOM (Ramírez et al., Reference Ramírez, Guhl, Messenger, Lewis, Montilla, Cucunuba and Llewellyn2012; Ramírez et al., Reference Ramírez, Tapia-Calle and Guhl2013) as an independent and discrete genotype of the TcI complex at least in Colombia and Venezuela (Figs 2–6), and indicate that TcI natural history is indeed complex and dynamic across geography and transmission cycles (Figs 3–5). This result was also recently suggested by network analysis at the country level (Gómez-Palacio et al., Reference Gómez-Palacio, Lopera, Rojas, Bedoya, Cantillo-Barraza, Marín-Suarez and Mejía-Jaramillo2016). Thus, the high phylogenetic incongruence among gene trees strongly indicates that gene flow occur through ecotopes (Figs 3–5). Consequently, different biological processes such as Intra Lineage Sorting (ILS) could be shaping the current parasite genetic structure, and this process is probably more widespread than previously thought (Ramírez and Llewellyn, Reference Ramírez and Llewellyn2014).
Recently, Zumaya-Estrada et al. proposed that TcIDOM originated in North/Central America before moving southwards and may be as ancient as humans in South America based on microsatellite data and concatenated clustering analyses (Zumaya-Estrada et al., Reference Zumaya-Estrada, Messenger, Lopez-Ordonez, Lewis, Flores-Lopez, Martínez-Ibarra and Llewellyn2012); these results are also consistent with those of Ramírez et al. (Reference Ramírez, Guhl, Messenger, Lewis, Montilla, Cucunuba and Llewellyn2012). However, methods that assume reciprocal monophyly, such as concatenation, may be biased by subjectivity (Hey, Reference Hey and Pinho2012); such methods are also bad estimators of real branch lengths in tree topologies and worse estimators of divergence times (Ogilvie et al., Reference Ogilvie, Bouckaert and Drummond2017). Our method herein employed for the first time in T. cruzi, supports the independence of TcIDOM; SL-IR and Cytb gene tree topologies illustrate that TcIDOM from Venezuela and Colombia are divergent (Figs 4 and 5), which could be accounted for by ILS and introgression. Indeed, our results indicate that, although TcIDOM is an emergent TcI genotype. Thus, a parsimonious explanation regarding these patterns in TcIDOM could be ‘divergence with gene flow,’ which has been demonstrated for a vast array of taxa [reviewed in (Shapiro et al., Reference Shapiro, Leducq and Mallet2016)]. In addition, human activity in the Anthropocene continuously changed the landscape, and potentially facilitated the dispersion of several parasitic strains among sylvatic and domestic cycles [e.g. (Lima et al., Reference Lima, Jansen, Messenger, Miles and Llewellyn2014; Poveda et al., Reference Poveda, Higuera, Urbano and Ramírez2017)]. However, one limitation of our study was the low number of loci and samples studied. Future studies should incorporate more loci and samples covering all the CD endemic range.
Moreover, the fact that TcIDOM is an emergent and discrete genotype of TcI adapted to domestic cycles with its own divergent evolutionary history (Fig. 6), implies different biological properties in this genotype. Recently, experimental infections in murine models (ICR-CD1/NIH and Balb-c mice) have demonstrated important differences in terms of parasitemia and tissue tropism as well as histopathological damage between Colombian TcIDOM strains and those sylvatic ones (TcI), in which TcIDOM seems to be less virulent (Cruz et al., Reference Cruz, Vivas, Montilla, Hernández, Flórez, Parra and Ramírez2015; León et al., Reference León, Montilla, Vanegas, Castillo, Parra and Ramírez2016). These experimental findings have also corroborated by evidence of histotropism between TcIDOM and sylvatic-like TcI strains in human patients with cardiomyopathy (Burgos et al., Reference Burgos, Diez, Vigliano, Bisio, Risso, Duffy and Schijman2010), supporting the current genetic subdivision.
Consequently, despite international consensus regarding Trypanosoma cruzi lineages or DTUs (TcI–VI) (Zingales et al., Reference Zingales, Miles, Campbell, Tibayrenc, Macedo, Teixeira and Sturm2012), and recent designation of TcBat as the seventh DTU (Marcili et al., Reference Marcili, Lima, Cavazzana, Junqueira, Veludo, Maia Da Silva and Teixeira2009; Lima et al., Reference Lima, Espinosa-Álvarez, Ortiz, Trejo-Varón, Carranza, Pinto, Serrano, Buck, Camargo and Teixeira2015), we did not think there was an optimal consensus method for identifying the differences and relationships within and between these major lineages for the described reasons above [reviewed in (Brenière et al., Reference Brenière, Waleckx and Barnabé2016)]. Additionally, division proposals are frequently contradictory [e.g. (Barnabé et al., Reference Barnabé, Mobarec, Jurado, Cortez and Brenière2016; Brisse et al., Reference Brisse, Barnabé, Bañuls, Sidibé, Noël and Tibayrenc1998; de Freitas et al., Reference de Freitas, Augusto-Pinto, Pimenta, Bastos-Rodrigues, Gonçalves, Teixeira and Pena2006; Flores-Lopez and Machado, Reference Flores-Lopez and Machado2011; Lewis et al., Reference Lewis, Llewellyn, Yeo, Acosta, Gaunt and Miles2011; Tomasini and Diosque, Reference Tomasini and Diosque2015)]. This issue is even more contentious at the intra-TcI level, where classifications have fluctuated among hosts and transmission cycles based on concatenated methods and single gene trees (Herrera et al., Reference Herrera, Bargues, Fajardo, Montilla, Triana, Vallejo and Guhl2007, Reference Herrera, Guhl, Falla, Fajardo, Montilla, Adolfo Vallejo and Bargues2009; Cura et al., Reference Cura, Mejía-Jaramillo, Duffy, Burgos, Rodriguero, Cardinal and Gürtler2010; Ramírez et al., Reference Ramírez, Duque and Guhl2011) until geography using multilocus data Llewellyn et al., Reference Llewellyn, Miles, Carrasco, Lewis, Yeo, Vargas, Torrico, Diosque, Valente, Valente and Gaunt2009b). Until recently, based on nuclear and mitochondrial concatenated MLST, classification of TcI has reduced to TcIDOM and sylvatic isolates, which indicates TcIDOM is an independently evolving lineage in the TcI complex (Ramírez et al., Reference Ramírez, Guhl, Messenger, Lewis, Montilla, Cucunuba and Llewellyn2012; Ramírez et al., Reference Ramírez, Tapia-Calle and Guhl2013).
A couple of studies have employed species delimitation approaches to clarify T. cruzi DTU relationships (Tomasini and Diosque, Reference Tomasini and Diosque2015) and examine the diversity of bat trypanosomes by Poisson tree processes (Cottontail et al., Reference Cottontail, Kalko, Cottontail, Wellinghausen, Tschapka, Perkins and Pinto2014). By employing current advances in Bayesian MSC (Ogilvie et al., Reference Ogilvie, Bouckaert and Drummond2017), our study represents the first comprehensive attempt to understand intra-lineage diversity and relationships in the TcI complex. Nevertheless, distinguishing between ILS and introgression is often challenging, and both are frequently confounded (Joly et al., Reference Joly, McLenachan and Lockhart2009). Here, we suggest that gene tree discordance may be influenced by these two processes. Consequently, it is important to note that StartBeast2 (Ogilvie et al., Reference Ogilvie, Bouckaert and Drummond2017) only accounts for ILS and not migration models. Moreover, in the face of complex T. cruzi population dynamics, future work that focuses on trypanosome biology and evolution must take into account lineage delimitation, conduct additional hypothesis testing, and further investigate if the patterns reflect introgression or ILS.
Based on random samples from Northern/Central America to South America and applying a Bayesian MSC approach, our results support the emergence of TcIDOM as an independently evolving genotype of the TcI complex at least in Colombia and Venezuela. In addition, we determined that neither geography nor hosts explain the gene tree discordance and genetic clustering among TcI strains (Introgression events). Other processes such as gene flow, divergence with gene flow, and ILS could shape the current parasite genetic structure and evolution.
However, the systematics, natural history, and epidemiology of T. cruzi are not fully understood. Here, we propose that combining MLST approaches with species coalescent-based methods for lineage delimitation of T. cruzi may be useful to prevent subjectivity and misidentification of parasite genetic structure that could be expanded to the other DTUs. Concatenation must be carefully used and taken into account in the light of the evaluation of hypotheses about the relationships between lineages and species.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182019000428.
Author ORCIDs
Juan David Ramírez González, 0000-0002-1344-9312.
Acknowledgments
We thank Mallory Eckstut, PhD, from Edanz Group (http://www.edanzediting.com/ac) for technical editing of an early version of this paper.
Financial support
This work was funded by DIRECCIÓN DE INVESTIGACIÓN E INNOVACIÓN from Universidad del Rosario.
Conflict of interest
None.
Ethical standards
Not applicable.