


INTRODUCTION
Schistosoma Weinland, 1858 is a genus of digenean (Strigeiformes: Schistosomatidae) blood flukes. Of its 21 known species, at least 7 are able to infect humans. Members of this genus cause the disease schistosomiasis and currently infect more than 200 million people in the tropics and subtropics (Chitsulo et al. Reference Chitsulo, Engels, Montresor and Savioli2000). Recently, much work has been directed at using DNA sequence-based studies to estimate a phylogeny for Schistosoma (see Snyder and Loker, Reference Snyder and Loker2000; Agatsuma et al. Reference Agatsuma, Iwagami, Liu, Rajapakse, Mondal, Kitikoon, Ambu, Agatsuma, Blair and Higuchi2002; Attwood et al. Reference Attwood, Upatham, Meng, Qiu and Southgate2002), assess the monophyly of major lineages (Lockyer et al. Reference Lockyer, Olson, Østergaard, Rollinson, Johnston, Attwood, Southgate, Horak, Snyder, Le, Agatsuma, McManus, Carmichael, Naem and Littlewood2003; Morgan et al. Reference Morgan, Dejong, Kazibwe, Mkoji and Loker2003; Webster et al. Reference Webster, Southgate and Littlewood2006) and interpret changes in mitochondrial gene order (Littlewood et al. Reference Littlewood, Lockyer, Webster, Johnston and Le2006). The current interest in Schistosoma phylogenetics arises from a desire to understand fully the relative roles of certain species of this genus in causing disease in humans (see Fenwick et al. Reference Fenwick, Rollinson and Southgate2006). In particular, there is a need to determine plesiomorphic character states (useful in phylogeographical studies) and to discern species and cohesive lineages within the genus. At least 1 of the 4 traditional species groupings within Schistosoma, namely the Schistosoma indicum-group, is now regarded as paraphyletic (Agatsuma et al. Reference Agatsuma, Iwagami, Liu, Rajapakse, Mondal, Kitikoon, Ambu, Agatsuma, Blair and Higuchi2002; Attwood et al. Reference Attwood, Upatham, Meng, Qiu and Southgate2002; Morgan et al. Reference Morgan, Dejong, Kazibwe, Mkoji and Loker2003; Webster et al. Reference Webster, Southgate and Littlewood2006). A reliable phylogeny for Schistosoma is important as false generalizations, based upon non-monophyletic (or artificial) groupings or on misidentified taxa, may lead to similarly incorrect conclusions about biology, epidemiology and disease control. A phylogeny is also required for incorporation into historical-biogeographical frameworks aimed at determining areas of origin and speciation as well as rates of speciation.
Schistosoma incognitum was first described by Chandler (Reference Chandler1926) from eggs found in freshly passed stools near a village in Krishnagar, Bengal, India. It is apparent that this taxon was rediscovered by Rao and Ayyar (Reference Rao and Ayyar1933), who described adult Schistosoma from pigs in Madras, India; the eggs of this taxon were identical to those of Chandler's paper. Rao and Ayyar named this taxon Schistosoma suis which was subsequently recognized as a synonym of S. incognitum (see Sinha and Srivastava, Reference Sinha and Srivastava1956). Traditionally S. incognitum has been placed within the S. indicum-group (see Rollinson and Southgate, Reference Rollinson, Southgate, Rollinson and Simpson1987), which also includes S. indicum, S. nasale and S. spindale; these 4 taxa have little in common and, aside from a South/Southeast Asian distribution, there appears to be no autapomorphy uniting them. These species differ in egg morphology and, although they can all be transmitted by Lymnaea snails, different species are used (Lymnaea luteola except S. spindale, Lymnaea acuminata, except S. indicum and S. incognitum). Similarly, all species except S. incognitum are known to be transmitted by Indoplanorbis exustus (see Combes et al. Reference Combes, Albaret, Arvy, Durette-Desset, Gabrion, Jourdane, Lambert, Leger, Maillard, Matricon, Nassi, Prevot, Richard and Theron1980). All of the recent phylogenetic studies on Schistosoma found S. incognitum to occupy a phylogentically basal position and to lie outside of a monophyletic ‘indicum clade’ comprised of the other 3 species (Agatsuma et al. Reference Agatsuma, Iwagami, Liu, Rajapakse, Mondal, Kitikoon, Ambu, Agatsuma, Blair and Higuchi2002; Attwood et al. Reference Attwood, Upatham, Meng, Qiu and Southgate2002; Morgan et al. Reference Morgan, Dejong, Kazibwe, Mkoji and Loker2003; Webster et al. Reference Webster, Southgate and Littlewood2006). However, the level of statistical confidence for the S. incognitum clade (strictly bi-partition) in these studies was low (7% to 67%). This information suggests some problem in resolving this part of the phylogeny. Further, to the extent of our knowledge, all of the studies to date have used the same S. indicum sample from Bangladesh, S. incognitum from the same population in northern Thailand and one S. nasale sample from Sri Lanka. For S. spindale, in the case of Lockyer et al. (Reference Lockyer, Olson, Østergaard, Rollinson, Johnston, Attwood, Southgate, Horak, Snyder, Le, Agatsuma, McManus, Carmichael, Naem and Littlewood2003), Morgan et al. (Reference Morgan, Dejong, Kazibwe, Mkoji and Loker2003), Webster et al. (Reference Webster, Southgate and Littlewood2006) a common Sri Lankan sample was used, but the remaining two studies (Agatsuma et al. Reference Agatsuma, Iwagami, Liu, Rajapakse, Mondal, Kitikoon, Ambu, Agatsuma, Blair and Higuchi2002; Attwood et al. Reference Attwood, Upatham, Meng, Qiu and Southgate2002) each used samples from unique populations in Thailand. Webster et al. (Reference Webster, Southgate and Littlewood2006) highlighted the need to investigate and understand the nature and extent of geographical variation at various genetic loci before these can be used in any ‘barcoding’ approach to taxonomy (see Hebert et al. Reference Hebert, Ratnasingham and Dewaard2003). The present work provides a population phylogeny for S. indicum-group taxa, the aim of which was to address these issues. Specifically, the work addresses the uncertainty regarding the position of S. incognitum in the Schistosoma phylogeny and the problem that previous studies had been mostly based on a small number of common population samples. The sampling of additional populations for 4 of the taxa and new loci for 11 was intended to screen for cryptic species and to improve phylogenetic resolution (following Zwickl and Hillis, Reference Zwickl and Hillis2002 and Pollock et al. Reference Pollock, Zwickl, Mcguire and Hillis2002). Taxonomic questions also exist in that there was some ambiguity regarding the original description of S. incognitum by Chandler and the origin of material. It is noteworthy that the egg of S. incognitum in Thailand (94–116 μm×45–63 μm length×width, spine 5–8 μm, our data) differs from that described by Chandler (Reference Chandler1926) for this taxon (95–100 μm×42–50 μm, spine 7–8 μm). Consequently, a population phylogenetic study was performed using independently collected samples, including new combinations of populations and loci in relation to earlier studies.
MATERIALS AND METHODS
Sampling
Samples were taken in 4 countries. Table 1 gives details of sampling sites and dates of collection. For PHS (see Table 1 for codes) populations of S. incognitum and S. spindale, adult worms were obtained following the methods described by Southgate et al. (Reference Southgate, Tchuem Tchuenté, Vercruysse and Jourdane1995) using the hamster (Mesocricetus auratus), as the laboratory definitive host, and cercariae from naturally infected snail intermediate hosts. Samples for the JKT and ZYL S. incognitum populations were obtained from field-trapped rodents by perfusion (Duvall and DeWitt, Reference Duvall and DeWitt1967). Samples of the other species/populations, which all commonly infect cattle, were collected from the livers and mesentery of slaughtered animals. Tegumental features (tubercles, spines, etc.), gross internal anatomy and egg morphology were used to identify the worms. DNA was preferentially extracted from females or separated worm pairs for which eggs had been observed and identified in corpo. Species identification followed Sinha and Srivastava (Reference Sinha and Srivastava1956) for S. incognitum, Montgomery (Reference Montgomery1906) and Rollinson and Southgate (Reference Rollinson, Southgate, Rollinson and Simpson1987) for S. spindale, and Rollinson and Southgate (Reference Rollinson, Southgate, Rollinson and Simpson1987) for S. indicum and S. nasale.
Table 1. Species sampled, collecting sites (populations) and dates for samples collected during the present study

* Indonesia.
DNA amplification and sequencing
DNA was extracted from single adult worms using a standard method (Winnepenninck et al. Reference Winnepenninck, Backeljau and De Wachter1993). Sequence variation was assessed at 2 loci, being partial sequences of the mitochondrial (mt) small and large ribosomal-RNA genes i.e. the rrnS and rrnL rRNA genes, respectively (gene notation following Le et al. Reference Le, Blair and McManus2000b). Sequences of the oligonucleotide primers used in the polymerase chain reaction (PCR) for the amplification of the rrnS locus are given in Attwood et al. (Reference Attwood, Upatham, Meng, Qiu and Southgate2002). The rrnS region amplified corresponded approximately to positions 11433–11760 in the complete mt genome sequence of S. spindale (see Littlewood et al. Reference Littlewood, Lockyer, Webster, Johnston and Le2006). Further details of the data set (including sample sizes and GenBank Accession numbers) are given in Table 2. A new primer pair was designed for the rrnL (F: TTGGTTGTGCTGACTACTCTG; R: GGCTTACACCGGTTTTAACT); the region amplified using this primer pair corresponded approximately to positions 10224–10851 on the same complete mt sequence. The efficiency of the PCR varied considerably between populations and, in some cases, this effect and the small number of worms available to us, led to a low number of replicates for some populations (see Table 2). In the case of S. nasale, there was insufficient material to obtain a readable sequence for this taxon at the rrnL locus.
Table 2. Summary of the DNA sequence data used in this paper
(Entries in bold denote populations or loci that were previously unstudied (or unpublished). Sequence lengths are in base pairs.)

+ Complete mt genome sequence in GenBank.
Two mt non-coding genes were also selected because, with their maternal pattern of inheritance, their smaller effective population size, the less expected correlation between mutations at different sites, and the more equal rates of substitution (i.e. no variation by codon), they were considered to represent potentially better recorders of phylogenetic events within a species group. In addition, the loci targeted were those within regions previously shown to exhibit ideal levels of variation in Schistosoma for this type of study (Attwood et al. Reference Attwood, Upatham, Meng, Qiu and Southgate2002), but which had not been previously studied for the populations involved here (thus providing new data).
Total genomic DNA was used as a template for PCR amplification on a Progene thermal cycler (MWG) employing standard PCR conditions (cfClackson et al. Reference Clackson, Güssow, Jones, McPherson, Quirke and Taylor1991). Unincorporated primers and nucleotides were removed from PCR products using the QIAQuick PCR purification kit (QIAGEN). Sequences were determined bidirectionally, directly from the products by thermal-cycle-sequencing using Big Dye fluorescent dye terminators and an ABI 377 automated sequencer (Perkin-Elmer), using procedures recommended by the manufacturers. DNA extracts were not pooled and one DNA sequence thus represented one worm. Sequences were assembled and aligned using Chromas (McCarthy, Reference McCarthy1996) and ClustalX (Thompson et al. Reference Thompson, Higgins and Gibson1994). DNA sequences for both strands were aligned and compared to verify accuracy. No-DNA template controls were included in all PCR runs in order to exclude any cross-over contamination.
Choice of substitution model and preparation of data
Consensus sequences for the populations sampled were grouped together into sets of aligned sequences of equal length (1 set for each locus), such that all taxa for which data were available were represented in each set (no rrnL sequence data were available for S. intercalatum or S. nasale) (Table 2). In addition, the rrnS and rrnL sequences for each population were concatenated and aligned to form a combined data set. No intrapopulation variation was found among the sequences. An outgroup sequence was taken from the GenBank (from NC002544) for S. japonicum from Leyte, Philippines (Le et al. Reference Le, Blair, Agatsuma, Iwagami, Humair, Campbell, Littlewood, Peacock, Johnston, Bartley, Rollinson, Herniou, Zarlenga and McManus2000a). For the rrnS data set only, a corresponding Paragonimus westermani sequence was included as an outgroup; no such data were available for the rrnL locus. An alignment of the sequences from other taxa of interest was attempted (e.g. Schistosoma haematobium), but no unambiguous alignment could be determined. Phylogenetic analysis was conducted using both a solely maximum likelihood (ML) approach and a Bayesian method (BM). The present data showed significant variation in the rate of substitution among sites, together with considerable bias among the 6 different types of nucleotide substitutions. In such cases, ML-based methods are considered more robust than most other commonly used phylogenetic methods, as they permit a better optimized model of substitution (Nei, Reference Nei, Miyamoto and Cracraft1991). The rrnS and rrnL data sets were analysed separately by ML and BM. The combined data set was analysed by BM only, because of the speed of BM and the ease of optimizing and applying models to each partition.
A suitable substitution model was selected using a hierarchical test of alternative models by mixed χ2-test as implemented in Modeltest v. 3.06 (Posada and Crandall, Reference Posada and Crandall1998). The Hasegawa, Kishino and Yano model, with estimates for among-site rate heterogeneity (HKY+G), was the model selected for the rrnS data. A transversional model, with allowance for the proportion of invariant sites was chosen for the rrnL data (TVM+I). The data were tested for substitution saturation using plots of the numbers of transitions and transversions against the ML genetic distance (following DeSalle et al. Reference Desalle, Freedman, Prager and Wilson1987). The indications of these plots were further evaluated using the entropy-based test (Xia et al. Reference Xia, Xie, Salemi, Chen and Wang2003) as found in the DAMBE (v. 4.5.29) software package (Xia, Reference Xia1999), which provides a statistical test for saturation. Statistics relating to polymorphism (see Table 3) were computed using the DNAsp (v. 3.51) (Rozas and Rozas, Reference Rozas and Rozas1999). The incongruence length-difference (ILD) test (Farris et al. Reference Farris, Kallersjo, Kluge and Bult1995), as implemented in PAUP* (v. 4.0b10; Swofford, Reference Swofford2002), was used to test for homogeneity between the rrnS and rrnL data partitions prior to combining them; the test was applied to informative sites only (following Lee, Reference Lee2001). In all analyses, gaps were treated as missing data and all characters were run unordered and equally weighted.
Table 3. Statistics relating to each data set used in the analyses
(Total number of sites in alignment L; number of taxa N; total number of sites excluding those with alignment gaps T; Jukes-Cantor corrected nucleotide diversity based on total number of mutations πJC; number of polymorphic sites with parsimony informative sites in parentheses PS; significance of Tajima's test for neutrality based on total number of mutations PT; θ per site based on the total number of mutations θ; significance of a likelihood ratio test for a molecular clock LRT.)

Phylogeny reconstruction: starting parameter values and priors
For the ML method heuristic searches were performed (under the respective model and starting parameters indicated by Modeltest) using PAUP* with random addition of sequences (10 replicates) and tree-bissection-reconnection branch swapping options in effect. Nodal support was assessed by bootstrap with 5000 replicates. Starting parameters for the BM were taken from Modeltest; these were then ‘optimized’ using a ML method with the Brent Powell algorithm in the phylogenetics software suite P4 (Foster, Reference Foster2004). The values from these optimizations were used as starting parameters for the first Bayesian analyses. A Metropolis-coupled Markov Chain Monte Carlo sampling process (mcmcmc, see Huelsenbeck et al. Reference Huelsenbeck, Ronquist and Hall2000) was used to search the parameter space of our evolutionary model and compute the posterior probability density.
Although a direct ML method was used in this study this was mainly to afford comparisons with earlier work. The final inferences were made using a BM; this is in accordance with a growing opinion that Bayesian phylogenetic analysis is not only faster in terms of computing time but also statistically superior to a solely ML method (Holder and Lewis, Reference Holder and Lewis2003). For example, such methods do not assume approximate normality or large sample sizes as would general ML methods (Van Dongen, Reference Van Dongen2006); they also allow the incorporation of prior information about the phylogenetic process into the analysis. In this study P4 was used to apply the BM; this employs the same method as MrBayes (Ronquist and Huelsenbeck, Reference Ronquist and Huelsenbeck2003) but allows consideration of unresolved trees (i.e. polytomies) and provides an automated (iterative) procedure for tuning the mcmc acceptance rates to acceptable levels. The mcmc was thereby tuned to give proposal acceptance rates between 10% and 70% for each data partition (this required over 5000 replicates). The P4 analyses (except for those using the polytomy prior) were repeated in MrBayes to reveal any topological disagreement.
The priors specified for the BM generally followed the default values found in MrBayes (3.1.2); a flat Dirichlet distribution was set as the prior for the state frequency and rate set priors (e.g. revmat, tratio) and the branch lengths were unconstrained. In contrast, a polytomy proposal was set as either zero (i.e., no favouring of multifurcations) or as e, e 2 or 10 to examine the effect this has on the posterior probabilities of the clades found; this implements a move (proposed by Lewis et al. Reference Lewis, Holder and Holsinger2005) to counter the problem of the spuriously high posterior clade probabilities returned by MrBayes relative to corresponding ML analyses. During the Bayesian analysis, model parameters and relative rates were set to be freely variable, so that, for the HKY+G model, there were 20 freely variable parameters and a total of 24 variable parameters; there were 4 discrete rate categories for the Γ-distribution.
Convergence of the mcmc was assessed by plotting split support (for the S. incognitum partition) for consensus trees over different generation time windows; the generation of convergence was considered to be that at which the support reached a plateau. In this way, a burnin of 200 000 generations was found to be adequate for all the analyses in this study. Posterior probabilities were then estimated over at least 500 000 generations beyond the assumed point of stationarity. Four simultaneous Markov chains were run (1 cold, 3 heated) and trees were sampled every 10 generations, 2 such runs were performed simultaneously. After 500 000 generations (post-stationarity) the average standard deviation of the split frequencies (between the 2 runs) was checked; the mcmc was considered complete if this SD was <0·01.
Hypothesis testing
Likelihood ratio tests (LRTs) were performed to assess the applicability of a molecular clock across the whole phylogeny (Felsenstein, Reference Felsenstein1988). The relative rates test of Nei and Kumar (Reference Nei and Kumar2000) was used to test the molecular clock hypothesis within individual clades (partitions) of the phylogeny. The tests were ML-based, using the HKY model, and tested among both constrained and unconstrained substitution models. In the case of the BM, the monophyly hypothesis was tested by determining the proportion of the trees in the posterior probability distribution consistent with a constraint tree containing only the monophyletic partition of interest; this gives the posterior-probability of the monophyletic bi-partition. For the ML method, the monophyly hypothesis was tested using the Shimodaira-Hasegawa test (SH-test, Shimodaira and Hasegawa, Reference Shimodaira and Hasegawa1999) as implemented in PAUP* with full optimization and 1000 bootstrap replicates to test between and among constrained and unconstrained topologies; the most parsimonious trees and the ML tree (unconstrained) were used as the set of trees for this comparison.
RESULTS
Sequence analysis
Table 3 provides basic statistics for the loci examined. The rrnL data appeared most informative having more parsimony informative polymorphic sites than the rrnS data. Of the aligned positions included in the analysis 42·6% were polymorphic in the rrnL data set (of these 64·0% were informative sites, the remaining 36·0% being singletons) and 48·0% in the rrnS set (of which 49·6% were informative). Plots of the numbers of transitions and transversions against genetic distance indicated slight substitution saturation at higher distances for the rrnL data. The test of Xia et al. (Reference Xia, Xie, Salemi, Chen and Wang2003) suggested that the levels of saturation were not likely to affect the analysis (Iss<Iss.c, P=0·0004 rrnL and 0·0001 rrnS, a lack of statistical significance here would imply a poor phylogenetic signal). Table 3 also shows that the nucleotide diversity (πJC) was greater in the rrnL data set. πJC for the combined data set was 0·180±0·017 but that for the S. incognitum clade alone was 0·035±0·010 and that for the S. spindale clade 0·020±0·006. In all cases the test of Tajima (Reference Tajima1989) failed to refute the hypothesis of neutral evolution. LRTs for the rrnS and rrnL data sets failed to support the hypothesis that the different lineages had been evolving at the same rate. However, the relative rates test for the S. spindale clade was not significant (P=0·05, both models constrained), suggesting that a molecular clock could be applied to this clade with caution. Substitutions within the S. incognitum clade were not clock like (P=0·005), unless transitions were excluded (P=0·104).
Phylogeny reconstruction
The topology for the optimal tree for the rrnS data by the ML method is given in Fig. 1A. The tree shows monophyletic clades for S. incognitum and S. spindale, but S. nasale is basal to a clade comprising S. intercalatum and all of the remaining S. indicum group taxa. Schistosoma indicum is at the root of the S. spindale clade and S. incognitum is at the root of the partition leading to S. intercalatum, S. indicum, S. spindale. Consequently, the traditional S. indicum group is paraphyletic in this phlylogeny. Schistosoma mansoni and S. japonicum are the two most basal taxa, with the latter placed closest to the outgroup. The ML tree for the rrnL data set and the BM tree for the combined (rrnS+rrnL) data set showed the same topology; that of the BM tree is shown in Fig. 1B. This phylogeny differs from that of the ML method in that the Bangladeshi population (not Indonesian) is basal in the S. incognitum clade, and S. japonicum and the S. incognitum clade form a trichotomy at the base of the tree. Levels of support for bipartitions on the BM tree (as posterior probabilities) were in all cases much higher than those of the rrnS ML phylogeny. Performing the BM with the polytomy prior turned off resulted in all probabilities being 100%, save for one at 98%, increasing the prior to e caused a slight drop in support, whereas further increases to e 2 and 10 had no further effect (Fig. 1B shows the clade support with the prior set to e 2). The topology and split support using MrBayes was almost identical to that returned by P4 with the polytomy prior turned off. The relationships within the S. incognitum clade on the ML tree were not well supported (occurring in only 75% of the trees sampled), whereas support for the alternative topology on the rrnL ML tree was 86% and on the BM tree 100%. Consequently, the relationships depicted by Fig. 1B are most likely to represent the true phylogeny.

Fig. 1. Phylograms based on different data sets used in the present study. (A) Maximum likelihood tree from the rrnS rRNA gene sequences only (outgroup Paragonimus westermani). Bootstrap support for each bi-partition is given as a percentage (of 5000 replicates) at each node. (B) Tree with maximum posterior probability for the combined rrnS and rrnL data set from a Bayesian method (outgroup Schistosoma japonicum). Numbers assigned to each node represent the posterior probability that the hypothesis represented by this bi-partition, and under all parameters of the model, is correct given the observed data.
Constraint analyses for the S. indicum group suggested a strong case for rejection of the hypothesis of monophyly. Full implementation of the SH-test on the rrnS data did not support the hypothesis of monophyly (−lnL unconstrained 1637·846, −lnL constrained 1651·254, P=0·032) as did the posterior probability of this constraint tree under the BM with the combined data set (P=0·025).
The average number of nucleotide substitutions per site (Dxy) was computed between the bi-partition at the root of each clade and the rest of the clade; this was used to provide a rough estimate of divergence time within both the S. incognitum and S. spindale clades. No correction for ancestral polymorphism was made because the Dxy within these clades was small (<0·02). Dejong et al. (Reference Dejong, Morgan, Wilson, Al-Jaser, Appleton, Coulibaly, Doenhoff, Haas, Idris, Magalhaes, Mone, Mouahid, Mubila, Pointier, Webster, Zanotti-Magalhaes, Paraense, Mkoji and Loker2003) hypothesized that the divergence of West African S. mansoni populations from those in East Africa was driven by the East to West colonization by Biomphalaria pfeifferi across Africa 135 000 years ago; this allowed estimation of a molecular clock rate for S. mansoni over a time range similar to that in the present study. On the basis of this rate, a 4% (divergence per million years) clock was assumed for the S. spindale clade and a 3% clock (adjusted following the transition ratio estimated by PAUP* to allow for the exclusion of transitions) for the S. incognitum clade. For the S. spindale clade (Bangladesh and Thai populations cf Sri Lanka) Dxy was 0·01957±0·01031 corresponding to a divergence time of 244 625±12 875 years before present (YBP). For the S. incognitum clade (Indonesian and Thai cf Bangladesh) Dxy was 0·01018±0·00528, corresponding to a divergence time of 165 420±85 797 YBP. It is noted that the ranges of these dates overlap and both taxa may have responded to the same driving event.
DISCUSSION
Interrelationships
The findings suggest that even using shorter sequences it is possible to recover a phylogeny with similar levels of statistical support and topology to those of studies based on much larger numbers of characters. The phylogeny shown in Fig. 1 generally agrees with that of Lockyer et al. (Reference Lockyer, Olson, Østergaard, Rollinson, Johnston, Attwood, Southgate, Horak, Snyder, Le, Agatsuma, McManus, Carmichael, Naem and Littlewood2003) and Webster et al. (Reference Webster, Southgate and Littlewood2006) which were based on extensive ssr DNA, lsr DNA and (mt) cox1 sequences and shared a common data set for the taxa considered here. The phylogenies presented here are based on a different data set but agree with those of the earlier studies in that S. indicum and S. spindale are shown to be sister taxa, that S. nasale is a relatively early divergent taxon showing affinities with African species (S. mansoni, S. interclatum) and S. incognitum, and that S. incognitum also diverges early in the phylogeny having close affinities with non-Asian taxa such as S. mansoni. The present results also provide strong evidence against the hypothesis of monophyly for the S. indicum group.
In spite of these similarities there are a few notable differences between the present findings and those of earlier work. In this study (Fig. 1A) S. nasale is placed at the root of a clade containing all other S. indicum group taxa and S. intercalatum, whereas Lockyer et al. (Reference Lockyer, Olson, Østergaard, Rollinson, Johnston, Attwood, Southgate, Horak, Snyder, Le, Agatsuma, McManus, Carmichael, Naem and Littlewood2003) found that S. intercalatum is just basal to S. nasale and S. incognitum is at the root of the clade containing the rest of the S. indicum group plus S. intercalatum and S. mansoni. The same basal position for S. incognitum is found in Fig. 1B (combined data set, BM) and this phylogeny shows good agreement with the two earlier studies. The reason for the basal position of S. nasale maybe that the rrnS contained fewer characters or that the taxon collected from Bangladesh and identified as S. nasale is not the same species as that from Sri Lanka used in the earlier studies. The phylogenies described by Lockyer et al. (Reference Lockyer, Olson, Østergaard, Rollinson, Johnston, Attwood, Southgate, Horak, Snyder, Le, Agatsuma, McManus, Carmichael, Naem and Littlewood2003) included 2 nuclear loci coding for rRNAs and this could also explain the difference in results. The paper of Agatsuma et al. (Reference Agatsuma, Iwagami, Liu, Rajapakse, Mondal, Kitikoon, Ambu, Agatsuma, Blair and Higuchi2002) and that of Attwood et al. (Reference Attwood, Upatham, Meng, Qiu and Southgate2002) both revealed differences in the phylogeny estimated from 28S rRNA sequences and from mt cox1 sequences. For example, the 28S phylogenies in both of the above papers placed S. haematobium within the S. indicum clade. In contrast, their rrnS and cox1 (mt) phylogenies agreed well with those of more recent studies. Also, the difference within the S. incognitum clade between the rrnS and rrnL-based phylogenies in the present study highlights the influence of locus choice on phylogenetic estimation. The differences can be due to recombination or concerted evolution (leading to a lack of phylogenetic signal) at the nuclear rDNA locus and/or incomplete lineage sorting at the mt loci. Such findings also have implications for the use of ‘barcoding’ in taxonomy. The kind of analysis employed in this study is expected to show a type I error rate of <3% at the sequence lengths used (Kolaczkowski and Thornton, Reference Kolaczkowski and Thornton2006). Lengthening the sequences (up to about 3000 bp) would theoretically lower this error rate even further and also the variance of certain parameter estimates; however, the extra benefit was considered to be too low to justify the extra cost, especially where sample material was limited and difficult to collect.
Analysis of the samples taken for the populations in this study did not reveal any obvious cryptic speciation and both S. spindale and S. incognitum formed cohesive groupings within which levels of genetic variation were typical of intraspecific comparisons. The nucleotide diversity was higher for the S. incognitum clade than for the S. spindale clade, but not so high as to signal the presence of any cryptic species. No sequence data for the Indian population of S. incognitum were available to this study and, as some morphological variation and perhaps host differences (none of the strains in this study was found to commonly infect pigs) have been found between Indian and Thai populations, additional work is required on this problem. The present study, however, permits us to state that the non-Indian S. incognitum populations all appear to be conspecific.
Phylogeography
The early divergent position of S. nasale and S. incognitum in the phylogeny and their close affinity with African species is consistent with the hypothesis of Barker and Blair (Reference Barker and Blair1996) ; that the S. indicum group arose after a reintroduction of Schistosoma to Asia, following an Asian to African migration and a radiation in Africa to produce the 3 main lineages of this genus (including S. mansoni). Agatsuma et al. (Reference Agatsuma, Iwagami, Liu, Rajapakse, Mondal, Kitikoon, Ambu, Agatsuma, Blair and Higuchi2002) and Attwood et al. (Reference Attwood, Upatham, Meng, Qiu and Southgate2002) noted the close affinity of Orientobilharzia turkestanicum Skrjabin, 1913 and S. incognitum and proposed that the latter probably represents the ancestral lineage in Asia from which the S. indicum group arose; however, the present result and that of Lockyer et al. (Reference Lockyer, Olson, Østergaard, Rollinson, Johnston, Attwood, Southgate, Horak, Snyder, Le, Agatsuma, McManus, Carmichael, Naem and Littlewood2003) show S. incognitum as basal to African taxa, suggesting that proto-S. incognitum arose and began to diverge in Africa or the Middle East. In this case the immediate ancestor of S. incognitum and Orientobilharzia in Africa must be assumed to be either extinct or as yet undiscovered in Africa. Schistosoma japonicum is at the root of the phylogeny in Fig. 1A; this suggests an Asian origin for Schistosoma as suggested by Snyder and Loker (Reference Snyder and Loker2000), with a subsequent radiation in the Middle East/Central Asia (now represented by S. incognitum and Orientobilharzia), next in Africa (from a proto-S. mansoni lineage), and finally in India and Southeast Asia (perhaps via S. nasale-like taxa) as the remainder of the S. indicum group. Attwood et al. (Reference Attwood, Upatham, Meng, Qiu and Southgate2002) noted that if one were to accept the idea of a re-colonization of Asia (from Africa) then one would expect the S. indicum group to be paraphyletic because either all ancestral (i.e. non-Asian) forms are now extinct or some of these taxa are represented by extant members of the S. indicum group. This aspect of the phylogeographical model is supported by empirical observation.
Times and triggers of phylogenesis in Asian Schistosoma
All Schistosoma restricted to rodents (or with rodents as their most significant definitive host) occur near the base of the Schistosoma phylogeny, whereas the species showing the more overall derived character states tend to be much more dependent upon Artiodactyla, particularly on Bovidae (cattle). The first murid rodents appear in the late Palaeocene fossil record (approximately 60 Ma (million years ago)); however, they do not appear in China until the late Miocene (around 15 Ma; Qiu and Li, Reference Qiu and Li2003). Consequently, if one assumes an Asian origin for Schistosoma with an antecedent from the S. sinensium lineage (which comprises mainly of rodent Schistosoma and includes S. japonicum), the origin of the genus should be mid-Miocene in the region of Tibet or Central Asia but prior to the major uplift (Fig. 2). Schistosoma incognitum maybe considered to have retained the plesiomorphic (rodent) host but Orientobilharzia probably represents an early off-shoot of the Asian rodent Schistosoma lineage that had evolved to utilize artiodactyls in Central Asia; this divergence would be expected following the Miocene radiation of mammals across Asia, Africa and Europe. A plausible route for Asian palaeo-Schistosoma into Africa would be during the great faunal exchange between Africa and Asia at the final closing of the Tethyan seaway (around 12 Ma). Such a date fits well with the conclusion of Despres et al. (Reference Despres, Imbert-Establet, Combes and Bonhomme1992) based on RFLPs, which suggested that S. mansoni was in Africa before the late Miocene. After a radiation in Africa involving ancestors of the S. mansoni and S. haematobium species groups (see Rollinson and Southgate, Reference Rollinson, Southgate, Rollinson and Simpson1987), antecedents of the S. indicum/spindale clade colonized India and the Sunda region. The date of this re-colonization is probably that of the Plio-Pleistocene (ca 2 Ma) large mammal migration from Africa to Asia along the Sinai-Levant dispersal tract (Matthee et al. Reference Matthee, Catzeflis and Douzery1997, Reference Matthee, Burzlaft, Taylor and Davis2001). The emergence of the Bovidae during the Pliocene and this migration of mammals are likely triggers for the divergence of the S. indicum clade from palaeo-S. incognitum or Orientobilharzia stock in Northeast Africa or Central Asia. Morgan et al. (Reference Morgan, Dejong, Kazibwe, Mkoji and Loker2003) suggested a role for the hippopotamus, infected by Schistosoma hippopotami Thurston, 1963 (a similarly early divergent taxon as Orientobilharzia), in the dispersal and divergence of Schistosoma at or before this time; however, as with Orientobilharzia, it is not clear whether this taxon originated in Africa or Asia.
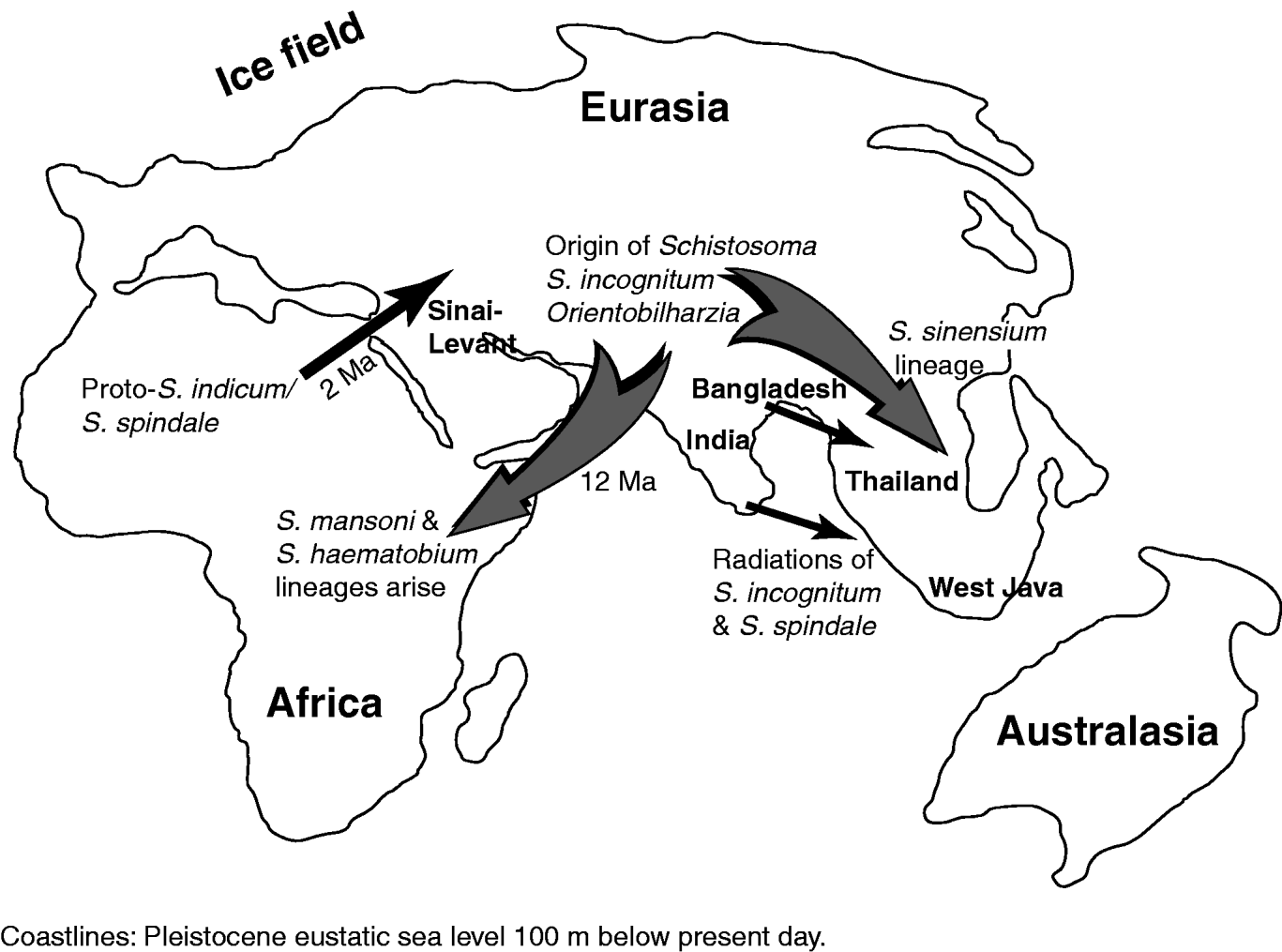
Fig. 2. Semi-schematic summarizing a phylogeography for Schistosoma. The genus is assumed to have arisen in rodents of Miocene Central Asia/Tibet (prior to major uplift there), with an antecedent typified by modern Schistosoma incognitum. The Schistosoma sinensium lineage next radiates in rodents in China and Southeast Asia. Orientobilharzia acquires artiodactyls as hosts in Central Asia; this lineage (or one based on proto-S. incognitum) enters Africa late Miocene, as the Tethyan seaway closes, to form the Schistosoma haematobium and Schistosoma mansoni lineages. The Pliocene large mammal radiation in Africa triggers the divergence of several lineages of Schistosoma utilizing Artiodactyla. The Plio-Pleistocene large mammal divergence into Asia (probably via the Sinai-Levant tract), and the emergence of Bovidae, then drives the divergence of a proto-Schistosoma indicum/Schistosoma spindale lineage from one of the African artiodactyle Schistosoma clades. Schistosoma of bovids establish on the Indian subcontinent and in Southeast Asia but apparently less so in Central Asia (possibly due to less speciation or many extinctions). In Southeast Asia both S. incognitum and S. spindale undergo near isochronous colonization and radiation from India/Bangladesh.
The present data indicated a divergence time ca 244 600 YBP for the Bangladesh and Thai populations of S. spindale from that of Sri Lanka; this is likely to be an over estimate because only 1 haplotype was used for the Sri Lankan population together with a minimum clock rate calibration (see Edwards and Beerli, Reference Edwards and Beerli2000). Intervals of very low sea level during Pleistocene interpluvials formed an intermittent exposed plain linking the present day Irrawaddy delta and the central Tenasserm range of Thailand to the Southeast. Consequently Thai and Bangladeshi populations of S. spindale may have been in contact throughout the Pleistocene, but it is more likely that the Thai population was founded within the last 10 000 years. The parasite was probably introduced via the movement of cattle from South Asia (including Bangladesh) into the Sunda region in association with human migration and trade. The divergence time estimated suggests that the Sri Lankan population was isolated by Pleistocene rises in sea level which broke the land bridge with India. A prolonged and extensive land bridge existed around 225 000 YBP and again at 165 000 YBP (Batchelor, Reference Batchelor1979). The assumed lack of extensive movement of cattle between Sri Lanka and Bangladesh during the Holocene may be a result of the expansion of the great Indian desert, and more recently to socio-political factors. Humans first developed a significant maritime technology 60 000 YBP (Bednarik, Reference Bednarik1995). Divergence within the S. incognitum clade (between the Sunda populations and Bangladesh) was dated at around 165 400 YBP. The present day definitive hosts of S. incognitum are murid rodents of rice fields and natural wetlands. Such taxa would have been well suited to dispersal across the Pleistocene wetlands of the exposed Sunda shelf which formed a land bridge between Thailand, the Malay Peninsula and West Java (around 170 000 YBP); this would have been irrigated by the West Sunda river and, to the north, the extended Chao Phrya (Tija, Reference Tija1980). The phylogeny in Fig. 1B is therefore consistent with a dispersal of S. incognitum out of Bangladesh (and India) to Sundaland (including Thailand and West Java) at a time of lowered sea level.
In summary, this work has further demonstrated the lack of monophyly for the S. indicum group and put forward explanations for this. The S. incognitum and S. spindale lineages in Southeast Asia have both been shown to form cohesive, monophyletic assemblages of con-specific taxa. The phylogeny estimated here lent support to the idea of an African origin for the lineage of those Schistosoma closely associated with Artiodactyla, followed by a Plio-Pleistocene introduction of a branch of this lineage into Asia (S. indicum, S. spindale). The divergences of S. incognitum and S. spindale populations appeared contingent on the same changes in sea level, but the two species were shown to have quite different phylogeographical patterns; this discrepancy may have arisen through differences in main definitive host type. The early divergent position of S. incognitum in the Schistosoma phylogeny was confirmed but future work is required to resolve its relationship with other basal taxa such as Orientobilharzia and S. mansoni. Extensive samples from the populations examined here (ideally based on other life-cycle stages), plus additional populations, are needed to enable the estimation of population genetic parameters and more accurate divergence times. Finally work is required on the taxonomy of S. incognitum, to compare Indian taxa with Southeast Asian in order to assess the significance of morphological variation and host differences noted in earlier studies.
This work was supported by Wellcome Trust Project grant (No. 068706) to S. W. A. Thanks are due to the staff of the Natural History Museum (NHM), London, for providing facilities for much of the work involved in this study, and in particular to Julia Llewellyn-Hughes and Claire Griffin for technical advice on automated DNA-sequencing, and to Peter Foster for advice on the application of P4.





