INTRODUCTION
Reconstructing the evolutionary history of species is essential to understand processes of evolution, and also to uncover and explain genetic diversity. It is proving particularly useful in the context of interacting and co-evolving species, such as host–parasite associations (Paterson and Banks, Reference Paterson and Banks2001). Co-phylogenetic methods (i.e. the comparison of independently constructed phylogenies) are now widely used to test hypotheses such as Farenholz's rule: ‘parasite phylogeny mirrors host phylogeny’ (Brooks, Reference Brooks1979; Klassen, Reference Klassen1992). This rule predicts that hosts and their parasites undergo strict co-speciation, producing congruent phylogenies [i.e. association by descent (Brooks, Reference Brooks1991)]. Although widely accepted in the early years of co-phylogenetic studies, Farenholz's rule and strict co-speciation are now seen as the exception rather than the rule, with host switching events playing a much larger role than previously assumed (de Vienne et al. Reference de Vienne, Refregier, Lopez-Villavicencio, Tellier, Hood and Giraud2013), resulting in complex associations between parasites and hosts (Hoberg and Brooks, Reference Hoberg and Brooks2008, Reference Hoberg and Brooks2015; Araujo et al. Reference Araujo, Braga, Brooks, Agosta, Hoberg, von Hartenthal and Boeger2015). Co-phylogenetic studies have to date been limited to only a few host–parasite model systems, and we are still a long way from understanding the factors constraining or promoting host range expansion in natural host–parasite associations.
Parasitic nematodes have received little attention from a co-phylogenetic perspective, yet they are an economically and ecologically important group (Hodda, Reference Hodda2011). Our study focuses on the host–parasite associations between New Zealand (NZ) lizards (lygosomine skinks and diplodactylid geckos) and their parasitic nematodes (Oxyurida), which inhabit the host's lower gut region. Skinks and geckos are two of the three terrestrial reptilian lineages found in NZ, Tuatara (Sphenodontidae) being the other. All skink and gecko species found in the wild are endemic with the exception of one invasive species, the rainbow skink (Lampropholis delicata), which arrived from Australia (Gill et al. Reference Gill, Bejakovtch and Whitaker2001). Both NZ skink and gecko lineages are monophyletic (Chapple et al. Reference Chapple, Ritchie and Daugherty2009; Nielsen et al. Reference Nielsen, Bauer, Jackman, Hitchmough and Daugherty2011). The ancestor of NZ skinks is thought to have arrived from New Caledonia approximately 16–22 million years ago (Hickson et al. Reference Hickson, Slack and Lockhart2000; Chapple et al. Reference Chapple, Ritchie and Daugherty2009) while evidence suggests that geckos arrived from Australia about 24 million years ago (Nielsen et al. Reference Nielsen, Bauer, Jackman, Hitchmough and Daugherty2011).
In comparison with their hosts, the nematodes parasitic in NZ lizards have received little attention in the literature. The most comprehensive study to date is a survey conducted by Ainsworth (Reference Ainsworth1992). Using morphological characteristics, the author identified and described two main species, Skrjabinodon trimorphi and Skrjabinodon poicilandri (Ainsworth, Reference Ainsworth1990). The former species was found only to infect skink hosts, while the latter only infected geckos. As members of Oxyuroidea, these nematodes are strictly monoxenous (direct, one-host life cycle) with the egg being the infective stage (Anderson, Reference Anderson2000). The host becomes infected when it accidentally ingests material contaminated with eggs (Anderson, Reference Anderson2000).
Our study aims to: (i) re-evaluate the diversity and taxonomy of nematodes parasitic in NZ skinks and geckos using molecular tools; (ii) to reconstruct the nematodes’ phylogenetic relationships; and (iii) to assess their co-phylogenetic patterns with their lizard hosts. Given the wide distribution of the parasites despite their limited dispersal ability, we hypothesize that there is more diversity present within NZ nematodes than has previously been detected by morphological examination, and that cryptic species are likely to be present. In terms of the co-evolutionary history between NZ lizards and their nematodes, our central hypothesis is that co-speciation will be the dominant mechanism in nematode–lizard associations where the parasites have a direct life cycle and the host has limited dispersal abilities. We also hypothesize that where host-switching events occurred, they involved hosts that share habitats and live in sympatry. The characteristics of this system (monophyletic hosts, large diversity of hosts, few non-native lizards present) make it an ideal model system for lizard–nematode associations with potential to advance our knowledge of evolutionary processes in parasites with direct life cycles.
MATERIALS AND METHODS
Nematode collection
A non-invasive nematode collection method was selected due to the conservation status of many of NZ's lizards. The method involved searching the host fecal pellets to recover any nematodes that had been expelled (Fenner et al. Reference Fenner, Godfrey and Bull2011; Jorge et al. Reference Jorge, Roca, Perera, Harris and Carretero2011; Gyawali et al. Reference Gyawali, Khanal and Shrestha2013). Fecal pellet collection took place over two sampling periods (December 2012–June 2013, and October 2013–May 2014), focused in the austral spring and summer months when lizard digestion rates are high. Pellets were collected either when the lizard released them spontaneously while being handled or using a method that involved gently massaging the lizard's abdomen to induce the expulsion of pellets from the intestines (Jorge et al. Reference Jorge, Carretero, Roca, Poulin and Perera2013a ). Permits for this exercise were obtained from the New Zealand Department of Conservation (permit numbers: 38672-FAU, 38674-FAU, 38678-FAU) and were approved by the University of Otago Animal Ethics Committee (protocol 36/14). All lizard species names used hereafter reflect the most recent classification, including informal names given to cryptic species yet to be formally described (Bell, Reference Bell2014).
Expelled pellets were collected in 1·5 mL Eppendorf tubes prefilled with 75–96% ethanol to preserve any nematodes. Each tube held feces from a single lizard. Samples were collected from wild populations, but also from captive populations when the species could not be sampled in the wild for conservation reasons. Avoiding samples from captive populations was important because captive animals may harbour nematodes that do not occur naturally in the locality from which they were collected. Such cases would obscure the true co-phylogeny patterns. For these reasons, samples from captive animals were only used if they were collected from wild-born animals kept in single-species colonies so that contamination was unlikely.
In total, 732 fecal samples were obtained from 28 lizard species (14 skink and 14 gecko species) (see Table S1 in Supplementary material) and from these samples 31 pellets were found to contain nematodes. In total, 46 nematodes were recovered (Table 1, Fig. 1). Where possible, three nematodes per locality/host species were selected for genetic analysis (specimens used in the genetic analysis are indicated in Table 1). A maximum of one nematode per fecal sample (1 per host individual) was used. Due to their size, female worms were always chosen over males for molecular analysis. Prior to genetic analysis, nematodes were photographed at a range of magnifications using an Olympus CX41 compound microscope with attached Olympus DP25 camera to provide a record of the nematodes that underwent genetic analysis. To photograph each nematode, a temporary slide was mounted using a 1:1 ratio mixture of glycerol and water (Foitová et al. Reference Foitová, Koubková, Baruš and Nurcahyo2008).

Fig. 1. Distributions of New Zealand skinks and geckos from which nematode parasites were recovered. Species abbreviations are as follows: O – Oligosoma, D – Dactylocnemis, N – Naultinus and W – Woodworthia, with a (c) indicating that samples were obtained from a captive population.
Table 1. Fecal samples from which parasitic nematodes were recovered and included in the genetic analysis

Samples in condition too poor to be used (i.e. only nematode cuticle/body wall remaining) are not listed. (C) indicates captive hosts, the location being their place of origin. GenBank accession numbers for sequences used in the genetic analysis are listed.
DNA extraction and sequencing
DNA extractions were performed using a PureLink® genomic DNA kit according to the manufacturer's instructions. Three partial gene fragments were selected for amplification, two nuclear genes: 18S rRNA (18S) and 28S rRNA (28S), and one mitochondrial gene: cytochrome oxidase subunit I (COI). The 18S fragment was amplified using the primers Nem 18S F and Nem 18S R designed by Floyd et al. (Reference Floyd, Rogers, Lambshead and Smith2005) and the 28S fragment with the primers 28S rD1·2a and 28S B described by Whiting (Reference Whiting2002). Due to difficulties associated with the amplification of the COI gene, new primers designed specifically for NZ pharyngodonid nematodes were used (SKR_F: 5′-TTT TTA TGG TGA TAC CTA TT-3′ and SKR_R: 5′-TAG TAT TAA AAT TAC GAT CAA-3′), which amplify approximately a 430 bp fragment.
All PCRs were performed in a total volume of 20 µL. This volume consisted of 4 µL of MyTaq™ Red reaction buffer [Bioline, Bioline (Aust) Pty, Alexandria, NSW, Australia], 1 µL of each primer at 0·5 mm, 0·2 µL of 0·4 mg mL−1 BSA (bovine serum albumin), 0·1 µL (0·5 U) of MyTaq™ DNA polymerase and 1·5–5 µL of DNA template. All reactions were performed on a Mastercycler pro S. Cycle conditions were typically as follows: denatured at 95 °C for 3 min followed by 35 iterations of: 40 s at 95 °C, 40 s at 47·6–54 °C (depending on primers), 1 min at 72 °C and ending with a final extension at 72 °C for 10 min. Amplified products were cleaned using ExoSAP-IT™ (USB, Cleveland, Ohio, USA) and sent to the Department of Anatomy at the University of Otago, Dunedin, New Zealand, for sequencing. Sequences were obtained for both directions using the same primers as in the PCR.
Phylogenetic analysis
All raw sequence files were imported into Geneious (version 8.0.3, Biomatters Ltd.) for analysis. Low-quality ends of sequencing reads were automatically trimmed (default parameters), and for each pair of sequence (forward and reverse) a contiguous sequence was assembled with the default parameters for the highest sensitivity and re-trim option. Read lengths (after trim) varied between 886 and 932 bp for 18S, 1114 and 1216 for 28S, and 369 and 682 for COI. Nematode contiguous sequences were aligned with the selected outgroups using the MAFFT algorithm (Katoh et al. Reference Katoh, Misawa, Kuma and Miyata2002) (algorithm setting E-INS-i for 18S and 28S, auto for COI, all other parameters default). Sequence quality (i.e. percentage of untrimmed bases of high quality) included in the alignments consisted of 99·21 ± 0·67 for 18S, 94·62 ± 5·07 for 28S and 96·33 ± 5·77 for COI (excluding GenBank sequences). The final alignments consisted of 764, 1156 and 369 bp for the 18S, 28S and COI genes, respectively.
Evolutionary models were estimated for each of the three amplified markers using a jModel test (jModelTest 2; Darriba et al. Reference Darriba, Taboada, Doallo and Posada2012) selecting for three substitution schemes and the BIC criterion, to restrict model selection to the ones implemented in MrBayes. Outgroups and all additional sequences for all analyses were obtained from GenBank (http://www.ncbi.nlm.nih.gov/genbank/) (Table 2). Phylogenetic analyses were performed for each dataset using Bayesian inference (BI). Bayesian analyses were performed in MrBayes 3·2·3 (Ronquist et al. Reference Ronquist, Teslenko, Van Der Mark, Ayres, Darling, Höhna, Larget, Liu, Suchard and Huelsenbeck2012) and run for 10 × 106 generations with random starting trees, sampling every 1000 generations. Two independent runs were performed each with one cold and three heated chains. The first 25% of the trees were discarded as burn-in and the remaining trees pooled. Mixing and convergence of each run was monitored by the statistics provided in MrBayes [values of standard deviation of partition frequencies (<0·01), potential scale reduction factors (PSRF) (1·00) and effective sample sizes (EES) (>200)]. A 50% majority-rule consensus tree was used to summarize the trees sampled from the post-burn-in trees.
Table 2. GenBank accession numbers for additional nematode sequences used in the phylogenetic analyses in this study

DS stands for direct submissions to GenBank. Species abbreviations are as follows: P: Parapharyngodon, Sp: Spauligodon, Ta: Thelandros, Te: Thelastoma.
Representative 18S sequences from all other available pharyngodonid genera (Parapharyngodon echinatus, P. cubensis, Spauligodon anolis, S. atlanticus, S. nicolauensis, Thelandros scleratus, T. tinerfensis) (Table 2) were selected to investigate the phylogenetic relationships of NZ parasitic nematodes. The species Thelastoma gueyei, which belongs to the same order but a different family, was used as an outgroup (Table 2). The jModel test revealed the best evolutionary model for this dataset to be SYM + I + G. The respective parameters were implemented in the phylogenetic analysis.
The species Thelandros tinerfensis and Paraphayngodon echinatus were selected as outgroups for the 28S and COI analyses. Available Spauligodon sequences were also included in the analysis to confirm the relationship between these species and the NZ lizard nematodes that was indicated in the 18S analysis. The model selected to analyse the 28S data was HKY + G. Prior to analysis, the COI alignments were translated to amino acids in order to determine the correct reading frame. To determine the best partition scheme and the respective models for COI, ParttitionFinder v1·1·1 (Lanfear et al. Reference Lanfear, Calcott, Ho and Guindon2012) was used. The best partition scheme consisted of two partitions: one combining the first and second codon position and another for the third codon position, with TrN + G and TIM + G as respective models. Finally, because there was no significant incongruence between the 28S and COI phylogeny, the 28S and COI genes were concatenated to obtain a more robust phylogeny. For the three specimens lacking the 28S gene fragment sequence information (Table 2), missing data was coded as ‘?’, since missing data are expected to have minor impact on the accuracy of phylogenetic analysis (Streicher et al. Reference Streicher, Schulte and Wiens2016). The best partition scheme selected by PartitionFinder v1·1·1 (Lanfear et al. Reference Lanfear, Calcott, Ho and Guindon2012) for the concatenated dataset, consisted of three partitions: 28S, COI first and second codon positions and COI third codon position, with the same model parameters as described above.
Genetic distances
To determine the genetic distances between and within each of the clades of NZ parasitic nematodes, and to the closest available related taxa (based on the combined 28S and COI inference trees; see ‘Results’ section), pairwise uncorrected p-distances were calculated in MEGA (version 6.06) (Tamura et al. Reference Tamura, Stecher, Peterson, Filipski and Kumar2013). Genetic distances were calculated separately for both the COI and 28S datasets.
Co-phylogeny analysis
To visualize host–parasite associations, a tanglegram was generated from the combined COI and 28S BI parasite tree and from the most complete available phylogenies for NZ geckos (Nielsen et al. Reference Nielsen, Bauer, Jackman, Hitchmough and Daugherty2011) and NZ skinks (Chapple et al. Reference Chapple, Ritchie and Daugherty2009) on the vector graphic software Inkscape (http://www.inkscape.org/). To explore the level of evolutionary congruence of each respective host–parasite association, the distance-based co-evolutionary analysis statistical tool PACo (Balbuena et al. Reference Balbuena, Míguez-Lozano and Blasco-Costa2013) was used. This analysis was appropriate because we did not have fully resolved phylogenies. The null hypothesis in this method is that the arrangement of the parasite phylogeny does not depend on the hosts (Balbuena et al. Reference Balbuena, Míguez-Lozano and Blasco-Costa2013). The PACo analysis was carried out in R Console version 3.1·2 (https://www.r-project.org/) implementing the associated script. Analysis was performed separately for gecko and skink parasites, using the COI dataset as parasite input. Host data input consisted of 16S partial sequences downloaded from GenBank (Table 3), representing each individual parasite and host link. Host sequences were aligned as for the nematodes (see above). Samples RP315 and RP317 were excluded from the gecko-Skrjabinodon analysis because of their captive origin. The congruence between the host and parasite phylogenies was measured with the residual sum of squares of the Procrustean fit, whose significance was established by 100 000 random permutations of the host–parasite association data. The contribution of each individual host–parasite association to the global fit was measured by means of jackknife estimation of their respective squared residuals, together with a 95% confidence interval associated with each host–parasite link.
Table 3. GenBank accession numbers for the 16S sequences obtained from NZ lizard species

All gecko sequences were obtained by Nielsen et al. (Reference Nielsen, Bauer, Jackman, Hitchmough and Daugherty2011) and skink sequences by Chapple et al. (Reference Chapple, Ritchie and Daugherty2009). Letters in () represent the location the host in this study came from: GB – Great Barrier Island, M – Maud Island, W – Wellington. Numbers in () represent the clade the O. polychroma species belonged to. Species abbreviations are as follows: O., Oligosoma; D., Dactylocnemis; N., Naultinus; W., Woodworthia.
RESULTS
Morphology
Morphological examination of the adult nematode specimens prior to genetic analysis revealed the presence of three distinct species according to available morphological descriptions. Two were assigned to the formally described species S. poicilandri (see Fig. S1 in Supplementary material) and S. trimorphi (see Fig. S2 in Supplementary material) (Ainsworth, Reference Ainsworth1990). All S. trimorphi specimens were found infecting skink hosts and all S. poicilandri specimens were found infecting gecko hosts. Note that a specimen of S. trimorphi (W7) from which we obtained DNA sequences comes from the type locality of the species, and three specimens of S. poicilandri (BH3, W1, W82) for which we obtained sequences come from near the type locality of that species (see Table 1). The third species was identified as the informally described species S. ‘five prong’ (sensu Ainsworth, Reference Ainsworth1992), which was found to be infecting Dactylocnemis pacificus on Great Barrier Island (Ainsworth, Reference Ainsworth1992). Although only one S. ‘five prong’ specimen was found and it was a juvenile specimen, it was easily assigned to this species because of the five large basal spines after which the species is named. Some specimens did not entirely match available morphological descriptions: GB10 had a greater body length, longer oesophagus, smaller tail size and fewer spines than S. poicilandri male morph 1. RP1688 was also larger in body size. However, genetic analysis suggests that there may be more diversity than morphological descriptions have previously identified (see below).
Genetic analysis
Of the 24 specimens that underwent genetic analysis, fragments for all markers were successfully obtained for 18 of the specimens. Unfortunately, nematode specimens found in two other skink species, namelly Oligosoma otagense and O. grande, both from Macraes Flat, were not successfully amplified, possibly due to DNA degradation.
Phylogenetic analysis
In both the 18S and 28S analyses, nematodes parasitizing skink hosts and those parasitizing gecko hosts grouped into two well-supported clades (0·89–1·0 posterior probability), but formed two paraphyletic groups (Fig. 2). Species of Skrjabinodon from skinks cluster together with species of Spauligodon, rather than with the gecko Skrjabinodon clade (Fig. 2). This relationship was also supported in the COI and concatenated analyses, where the nematodes from skinks formed a monophyletic clade with Spauligodon species (Fig. 3). From this analysis, six lineages were identified for the gecko parasites, and three for the skink parasites (Fig. 3). For the gecko parasites, each lineage contained specimens collected from the same host species and location, or nearby locations as with sample RP84 from Maud Island, which formed a lineage with the Wellington specimens. The exception was lineage 5 where the two specimens came from different hosts, but these hosts were both captive animals at the same location. Although the concatenated 28S and COI phylogeny was able to better resolve between-lineage relationships than the other analyses, they were not always well resolved (Fig. 3). An interesting feature of the inferred Skrjabinodon phylogeny is the high divergence of specimen RP999 (D. pacificus, Great Barrier Island), which was morphologically identified and Skrjabinodon ‘five prong’, from the rest of the specimens (posterior probability 1·0).

Fig. 2. The 18S inference tree showing the phylogenetic relationships of the parasitic nematodes recovered from New Zealand gecko and skink species (shaded in grey). The tree includes representatives from the order Oxyurida. The values at nodes represent posterior probabilities (values below 75% are not shown).

Fig. 3. Inference tree for the concatenated 28S gene and COI gene data obtained from parasitic nematodes that were recovered from New Zealand gecko and skink species (shaded in grey). The values at nodes represent posterior probabilities (values below 75% are not shown). Numbered groupings show lineages identified by this tree (G for gecko parasites and S for skink parasites). Symbols show the localities at which the nematodes were recovered from their hosts.
Genetic distances
As inferred in Bayesian analyses, nematodes infecting NZ skinks are genetically more similar to species of Spauligodon than to those from geckos. For the 18S, the nematodes recovered from gecko hosts had a genetic distance of 4·7% to the group formed by species of Spauligodon and the ones from NZ skink hosts. Nematodes found infecting the NZ skinks showed 1·9% genetic differentiation (uncorrected p-distance) from species of Spauligodon. The same pattern was recovered for the COI and for 28S, where NZ skink parasites were less divergent from Spauligodon taxa than from the other Skrjabinodon-infecting NZ geckos (COI: 24·8 vs 20·6% uncorrected p-distance; 28S: 20·4 vs 9% uncorrected p-distance). Within groups, the COI marker was highly divergent between nematode lineages from gecko hosts (Table 4). The largest between-group genetic distances were found between the nematode morphologically identified as S. ‘five prong’ (host D. pacificus), and the other five lineages, which ranged between 24·8 and 27·5% (uncorrected p-distance) for COI and an average of 6·7% for 28S. Between parasites from skink lineages, distances based on the COI marker ranged from 10 to 12·5% (uncorrected p-distances; Table 5), whereas for the 28S marker, distances ranged from 0·9 to 2·2% (uncorrected p-distances, Table 5).
Table 4. Pairwise uncorrelated p-distances between clades for New Zealand gecko nematodes

Lineages were determined based on the joint 28S–COI analysis (see Fig. 3 for additional details).
Table 5. Pairwise uncorrelated p-distances between clades for nematodes obtained from New Zealand skink species
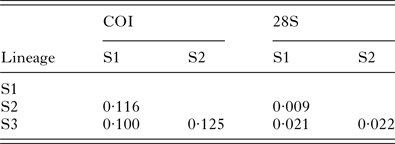
Lineages were determined based on the joint 28S–COI analysis (see Fig. 3 for details).
Co-phylogenetic analysis
The contribution of each parasite–host link to the global fit between host and parasite phylogenies can be visualized in Fig. 4. The PACo analysis provided clear support for the overall congruence between Skrjabinodon species and their NZ gecko hosts (m 2 XY = 0·0043, P < 0·00001). The residual bars in Fig. 4B show that the associations involving Woodworthia cf. brunnea contribute little to m 2 XY and, therefore, likely represent co-evolutionary links. Similarly, the majority of the Woodworthia maculata and W. ‘Otago large’ associations had low squared residuals but the confidence intervals were large, crossing the median squared residual value, making it difficult to assess their contribution to co-phylogenetic patterns. The PACo analysis for the NZ skink hosts and their associated nematode lineages yielded an m 2 XY of 0·0014 with a permutational value of P < 0·0735. Thus, there was no evidence for an overall congruence between NZ skink host and parasite associations.

Fig. 4. Tanglegram of the cophylogenetic relationships between NZ Skrjabinodon parasites and their gecko hosts (A) and skink hosts (C). Gecko host tree adapted from Nielsen et al. (Reference Nielsen, Bauer, Jackman, Hitchmough and Daugherty2011) and skink host tree adapted from Chapple et al. (Reference Chapple, Ritchie and Daugherty2009). Parasite tree adapted from the concatenated 28S gene and COI gene data inference tree. (B) and (D): Jackknife residual bars resulting from PACo analysis for each respective host–parasite association represented in the tanglegram (except for captive specimens). The dotted line represents the mean residual value and the error bars are the upper 95% confidence intervals. Host–parasite links with bars below the mean residual value are considered indicative of co-speciation links.
DISCUSSION
The aim of this study was to re-evaluate the diversity and taxonomy of nematodes parasitic in NZ lizards, to estimate their phylogenetic relationships and to assess their co-phylogenetic patterns. The following discussion addresses each of these issues in turn.
Taxonomy
Prior to this study, the classification of nematodes parasitic in NZ lizards was solely based on morphological characters (Ainsworth, Reference Ainsworth1985, Reference Ainsworth1990, Reference Ainsworth1992). To date, Ainsworth's (Reference Ainsworth1992) detailed morphological survey of nematodes parasitizing native lizards remains the most comprehensive taxonomic study. She identified all recovered nematodes to the family Pharyngodonidae, which agreed with several other authors who had previously recovered nematodes from NZ reptiles (Barwick, Reference Barwick1959; Clark, Reference Clark and Alison1982; Ainsworth, Reference Ainsworth1985). At the level of genus, the same study assigned pharyngodonid nematodes of NZ skinks to the genus Skrjabinodon (Ainsworth, Reference Ainsworth1990, Reference Ainsworth1992). While genetic data from our study confirm that nematodes parasitic in both NZ skinks and geckos belong to the family Pharyngodonidae, they do not form a monophyletic group. In fact, nematodes infecting NZ skinks are genetically more closely related to Spauligodon than to NZ Skrjabinodon species infecting NZ geckos (Figs 2 and 3).
Both S. trimorphi and S. poicilandri were described by Ainsworth (Reference Ainsworth1990) from the Wellington region, infecting O. polychroma skinks and W. maculata geckos, respectively. In our study, we also sampled parasites from the same hosts and locality, which also morphologically aligned with descriptions provided by Ainsworth (Reference Ainsworth1990, Reference Ainsworth1992) suggesting that the same ‘species’ were examined. Incongruence between morphological and molecular data has already been reported for pharyngodonid nematodes (Jorge et al. Reference Jorge, Perera, Roca, Carretero, Harris and Poulin2014). However, the fact that molecular data showed that the Skrjabinodon species parasitizing NZ lizards are paraphyletic highlights the importance of a proper assessment of how morphological characters reflect the relatedness between species and genera, and how they have evolved in this family.
For the NZ gecko nematodes, this study confirms that all specimens here belong to the same genus. Morphologically they fit into the genus Skrjabinodon, as determined by Ainsworth (Reference Ainsworth1992). As there are currently no other available sequences for species of Skrjabinodon, the relationship of NZ specimens to other Skrjabinodon species could not be examined. However, the NZ specimens did not group with the other genera included in this analysis, supporting Ainsworth (Reference Ainsworth1990) whose survey suggests that this is the only genus present within NZ geckos. Comparing NZ Skrjabinodon species to other congeners would allow the monophyly of this genus to be assessed.
Phylogeny, diversity and cryptic species
Our second aim was to estimate the phylogenetic relationships of the nematodes parasitic in NZ skinks and geckos. Due to the small number of specimens recovered and the high host diversity, phylogenetic relationships of NZ Skrjabinodon parasites may not be representative of the actual parasite diversification (Fig. 4). On a finer level, several conclusions can be drawn about NZ Skrjabinodon species. First, there is greater genetic diversity present within these nematodes than has previously been detected; nematodes from each location grouped into individual lineages and present high genetic differentiation. For example, at the COI marker the smallest distance between Skrjabinodon lineages infecting geckos was over 16% (see Table 4). These distances are comparable to interspecific distances obtained for other nematode species within the same genus such as between Spauligodon occidentalis and S. atlanticus (12·8%) (Jorge et al. Reference Jorge, Roca, Perera, Harris and Carretero2011, Reference Jorge, Perera, Carretero, James Harris and Roca2013b ), or within the genera Oesophagostomum (11·5–13·7%) (de Gruijter et al. Reference de Gruijter, Polderman, Zhu and Gasser2002), Ancylostoma (4·8–11·1%) (Hu et al. Reference Hu, Chilton, Zhu and Gasser2002) and Pellioditis (Derycke et al. Reference Derycke, Remerie, Vierstraete, Backeljau, Vanfleteren, Vincx and Moens2005). Blouin et al. (Reference Blouin, Yowell, Courtney and Dame1998) suggested that if two mtDNA sequences differ by 10%, we should consider whether these are truly conspecifics. These data give us a snapshot of the diversity present within these nematodes and indicate that they may contain as much diversity as their hosts (Fig. 4). As with their hosts (Nielsen et al. Reference Nielsen, Bauer, Jackman, Hitchmough and Daugherty2011), we may expect cryptic diversity within each NZ parasite clade. Cryptic species are common within parasitic nematodes (Grillo et al. Reference Grillo, Jackson, Cabaret and Gilleard2007; Tan et al. Reference Tan, Chilton, Huby-Chilton, Jex, Gasser and Beveridge2012; Karpiej et al. Reference Karpiej, Dzido, Rokicki and Kijewska2013) and the COI gene has been highly useful for their identification because of its fast mutation rate (Ballard and Whitlock, Reference Ballard and Whitlock2004; Frézal and Leblois, Reference Frézal and Leblois2008). Ainsworth (Reference Ainsworth1990) rejected the possibility of cryptic species within S. trimorphi and S. poicilandri. However, her study was based on allozyme electrophoresis from specimens from single host species, from one locality. We present molecular data from a wider range of hosts and localities across NZ. Nevertheless, only a few specimens were found, preventing a proper morphological study to assess the cryptic nature of these nematodes. Further, genes do not necessarily reflect the evolution of species (Szöllosi et al. Reference Szöllosi, Tannier, Daubin and Boussau2013), and multiple genes should be used to infer species delimitation (Dasmahapatra et al. Reference Dasmahapatra, Elias, Hill, Hoffman and Mallet2010; Fujita et al. Reference Fujita, Leaché, Burbrink, Mcguire and Moritz2012; Collins and Cruickshank, Reference Collins and Cruickshank2013). For this reason, we only go as far as calling the genetically diverse groups we uncovered provisionally cryptic.
Within lineages, nematodes from sympatric hosts of the same species always grouped within the same clade and had relatively low genetic distances comparable with those found intra-specifically in other nematodes (de Gruijter et al. Reference de Gruijter, Polderman, Zhu and Gasser2002; Hu et al. Reference Hu, Chilton, Zhu and Gasser2002). This suggests that only one ‘species’ per host/locality was detected. The exception was Skrjabinodon specimens collected from W. ‘Otago large’ at Macraes Flat. At the COI marker the sample RP330 had a distance of 15% (uncorrected p-distance) to the other samples from that area (RP945 and RP1377), which is comparable with interspecific distances and suggests that there are potentially two nematode species parasitizing this host in that locality.
Another point of interest relates to the S. ‘five prong’ lineage, which was basal to all other NZ gecko Skrjabinodon lineages. However, this does not indicate that this lineage represents a basal divergence in the evolutionary history of NZ gecko Skrjabinodon species. It should be noted that the basal position of this species and its relationship with the other gecko nematodes do not agree with the phylogenetic relationship of its respective host, since Dactylocnemis is more closely related to Naultinus than to Woodworthia (Nielsen et al. Reference Nielsen, Bauer, Jackman, Hitchmough and Daugherty2011; Fig. 4A). These differences are well evidenced by the high squared residual value obtained for this host–parasite link (Fig. 4B). Nevertheless, the data show that this specimen is genetically very different from all others, supporting Ainsworth's (Reference Ainsworth1992) conclusion that S. ‘five prong’ is indeed a different species requiring formal description. Interestingly, Skrjabinodon species with three large basal body spines are limited to NZ geckos and three other gecko species from Australia (Jones, Reference Jones2013), where the ancestor of NZ geckos is thought to have originated (Nielsen et al. Reference Nielsen, Bauer, Jackman, Hitchmough and Daugherty2011). This could indicate that Skrjabinodon arrived in NZ with the colonization of the host and three base ‘prongs’ is an ancestral state. Further sampling needs to be conducted to uncover the evolutionary history of these species.
Few nematode specimens were recovered from NZ skinks despite similar numbers of fecal samples to geckos, most likely indicating lower infection prevalence in skinks (Ainsworth, Reference Ainsworth1992). The combined 28S and COI phylogenetic analysis had very good general support, although for better phylogenetic accuracy a much wider taxon sampling is needed. The pairwise genetic distances at the COI marker were lower for NZ skink Skrjabinodon species despite similar geographical distances (i.e.: highest genetic distance between skink nematodes = 12%, while lowest between clade distance for gecko Skrjabinodon species = 17%, uncorrected p-distances). This could reflect differences in the evolution of these species, i.e. populations of nematodes infecting skinks may have remained connected for longer periods and therefore diverged later than those of gecko Skrjabinodon. Alternatively, differences in host ecology may influence the connectedness of parasite populations. For example, NZ gecko species tend not to co-exist and/or occupy different altitudes, habitats or niches (Jewell, Reference Jewell2006). In contrast, skinks more readily occupy the same habitat and several species occur at the same site (Jewell, Reference Jewell2006).
Co-phylogeny
The primary motivation behind this study was to explore the co-phylogenetic patterns between NZ lizards and their nematode parasites. In particular, the main aim of this study was to determine which process, host switching or co-speciation, has been the most common path to modern day associations between NZ lizards and their parasitic nematodes. The working hypothesis was: (i) that co-speciation played a major role in the evolution of associations between NZ skinks and geckos and their nematodes, and (ii) that where host-switching events occurred they only took place between sympatric hosts that share habitats. In terms of the relationship between the skink hosts and their associated nematodes, the skink phylogeny did not significantly predict the ordination of parasitic nematodes. However, our study did not cover all of the nematode diversity and overall conclusions about the co-phylogenetic patterns remain tentative, therefore we could not reject the null hypotheses with the current dataset.
In contrast, the co-phylogenetic analysis of Skrjabinodon species and their gecko hosts revealed that the host phylogeny significantly predicted the parasite phylogeny. This result indicates that co-speciation may have played an important role in the evolution of NZ gecko and Skrjabinodon associations. However, congruent topologies can also be obtained by host-switching to a closely related host followed by speciation (de Vienne et al. Reference de Vienne, Refregier, Lopez-Villavicencio, Tellier, Hood and Giraud2013). Furthermore, not all host–parasite associations supported a congruent host–parasite phylogenetic history (see Fig. 4), suggesting that other events such as host switching have also played an important, although perhaps smaller role. The macro-evolutionary history of Skrjabinodon infecting NZ lizards cannot be fully reconstructed at this stage due to our limited sample size. It is important for determining the overall co-phylogeny that nematodes from a greater range of hosts and locations are added to this dataset. To properly assess the occurrence of co-speciation it is also fundamental to demonstrate identical ages for each node of host and parasite phylogenies (de Vienne et al. Reference de Vienne, Refregier, Lopez-Villavicencio, Tellier, Hood and Giraud2013). The lack of fossils or appropriate calibrations prevents us from producing a time-calibrated phylogeny for the NZ nematodes.
Additional data is also vital to testing the second part of our hypothesis that host switches should only occur between sympatric hosts. Investigating members of the Woodworthia clade that occur in sympatry would be particularly interesting. It is recognized that host switches are more likely to occur between species that are closely related (Jackson, Reference Jackson1999). Thus, this complex is primed for host switching events. Could this explain the large genetic diversity found between nematodes that came from W. ‘Otago large’ geckos from the same area? While no definitive conclusions about the co-phylogenetic patterns can be drawn from the current data, it is clear that Fahrenholz's rule is too simple to explain the history between hosts and their parasites.
In conclusions, this research expands knowledge of the native nematodes parasitizing NZ reptiles. Importantly, this work has revealed that nematodes parasitizing NZ skinks are genetically more closely related to Spauligodon than to the other Skrjabinodon species infecting NZ geckos. However, most nematodes obtained from geckos came from broad-toed gecko species. To better understand the overall co-phylogenetic patterns between geckos and their Skrjabinodon species, ideally parasites would be obtained from all NZ gecko clades. Our results also highlight that there is much more diversity within these nematodes than previously recognized, potentially as much diversity as exists within the reptile hosts. We provide evidence for provisionally cryptic species; further sampling of these nematodes is required to uncover their full diversity. It is important to understand their biodiversity so conservation managers can make informed decisions on how best to manage both parasite and host populations.
SUPPLEMENTARY MATERIAL
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182016002365.
ACKNOWLEDGEMENTS
We thank the following for contributing samples to this study: Marleen Baling, Ben Barr, Bernie Buhler, Luca Butikofer, Hermann Frank, Sophie Gibson, Jordi Janssen, Ngaire Jury, Dennis Keall, Linda Kilduff, Karen Ludwig, Les Moran, Sophie Penniket, James Reardon, Anita Spencer, Cindy van Veen, Barbara Watkins, Chris Wedding and members of EcoGecko Consultants: Carey Knox, Sabine Melzer, Rachel Innes and Sarah Herbert.
FINANCIAL SUPPORT
This study was funded internally by the Zoology Department, University of Otago.