


INTRODUCTION
Geographical patterns in the distribution of parasitic organisms can provide insights into the ecological and evolutionary forces that determine host-parasite associations. Regional parasite fauna are shaped by local colonization and extinction events, which in turn may be driven by a combination of host immune responses, competition between parasites, and stochastic changes in the environment (Apanius et al. Reference Apanius, Yorinks, Bermingham and Ricklefs2000; Fallon et al. Reference Fallon, Bermingham and Ricklefs2003, Reference Fallon, Bermingham and Ricklefs2005; Ishtiaq et al. Reference Ishtiaq, Clegg, Phillimore, Black, Owens and Sheldon2010). Parasites with a direct life cycle are often widespread in their geographical distribution. Their presence is dependent primarily on the occurrence of a definitive host. Vector-mediated parasites such as Plasmodium spp. and Haemoproteus spp. rely on the presence of both an appropriate host and a competent vector (Bennett et al. Reference Bennett, Campbell and Cameron1974; Apanius et al. Reference Apanius, Yorinks, Bermingham and Ricklefs2000). The primary vectors for Haemoproteus spp. are known to be biting midges of the genus Culicoides (Diptera: Ceratopogonidae) and louse flies (Diptera: Hippoboscidae) (Atkinson and van Riper, Reference Atkinson, van Riper, Loye and Zuk1991). Species of avian Plasmodium are most commonly transmitted by mosquitoes (Valkiũnas, Reference Valkiũnas2005). Parasites with specialized vector associations tend to have a restricted range of hosts (Killick-Kendrick, Reference Killick-Kendrick, Killick-Kendrick and Peters1978) and consequently have restricted geographical distributions (Lajeuness and Forbes, Reference Lajeunesse and Forbes2002). In contrast, the use of a broad spectrum of blood-feeding vectors may facilitate host switching in generalist parasites (Githeko et al. Reference Githeko, Service, Mbogo, Atieli and Juma1994), enlarging the geographical range of those parasites. Furthermore, the spatial distribution of parasites is closely linked to the environmental, ecological, climatic and geographical connectivity which governs the faunistic exchange between populations.
Oceanic archipelagos have been used as natural laboratories for understanding the evolutionary processes of speciation and divergence (Mayr, Reference Mayr1942; MacArthur and Wilson, Reference MacArthur and Wilson1967) and provide a unique opportunity to explore community assemblage in similar yet geographically discrete units. The western Indian Ocean region has been used in recent years as a model area to distinguish between patterns of vicariance and dispersal in various taxonomic groups (Bossuyt and Milinkovitch, Reference Bossuyt and Milinkovitch2001; Biju and Bossuyt, Reference Biju and Bossuyt2003; Renner, Reference Renner2004a,Reference Rennerb), and to understand patterns of island colonization from neighbouring continents (e.g. Warren et al. Reference Warren, Bermingham, Bowie, Prys-Jones and Thebaud2003, Reference Warren, Bermingham, Prys-Jones and Thebaud2005; Fig. 1). The western Indian Ocean region is an avian diversity hotspot with nearly 300 bird species, of which nearly 60% are endemic species. Over 55 endemic species are endangered or threatened by their restricted range. Given the documented and suspected threats posed by avian blood parasites in other island birds (Beadell et al. Reference Beadell, Ishtiaq, Covas, Melo, Warren, Atkinson, Bensch, Graves, Jhala, Peirce, Rahmani, Fonseca and Fleischer2006; Matson and Beadell, Reference Matson, Beadell, Morand and Krasnov2010), haematozoa remain surprisingly understudied in the Indian Ocean region.
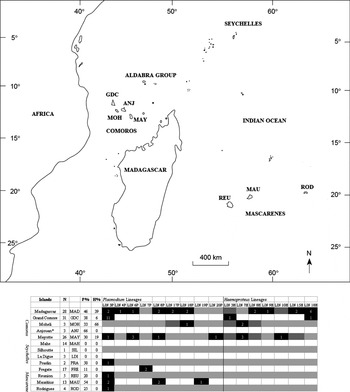
Fig. 1. Distribution of unique Plasmodium and Haemoproteus lineages by island. MAD=Madagascar; GDC=Grand Comore; MOH=Moheli; ANJ*=Anjouan (no lineage information available); MAY=Mayotte; MAH=Mahe; SIL=Silhoutte; LDI=La Digue; PRA=Praslin; FRE=Fregate; REU=Reunion; MAU=Mauritius; ROD: Rodrigues. P%=Plasmodium prevalence, H%=Haemoproteus prevalence. Total parasite prevalence includes 91 bp sequences (not used in phylogenetic analysis).
Whilst early microscopy-based surveys of Indian Ocean avifauna revealed a diverse array of blood parasites (Lowery, Reference Lowery1971; Peirce et al. Reference Peirce, Cheke and Cheke1977; Bennett and Blancou, Reference Bennett, Campbell and Cameron1974; Savage, Reference Savage2003; Savage et al. Reference Savage, Robert, Goodman, Raharimanga, Raherilalao, Andrianarimisa, Ariey and Greiner2009), they have so far been little-explored using molecular techniques, and their diversity and distribution is poorly known relative to that of the neighbouring African continent (e.g. Bensch et al. Reference Bensch, Stjernman, Hasselquist, Östman, Hansson, Westerdahl and Pinheiro2000; Beadell et al. Reference Beadell, Covas, Gebhard, Ishtiaq, Melo, Schmidt, Perkins, Graves and Fleischer2009). Given the long-term geographical isolation and concurrent evolution of a unique avifauna with few migrants, the evolution of indigenous parasite fauna with endemic hosts could be considered as a possibility. With the recent advent of molecular techniques, a wealth of genetic diversity in parasite lineages has been revealed (Bensch et al. Reference Bensch, Stjernman, Hasselquist, Östman, Hansson, Westerdahl and Pinheiro2000; Ricklefs and Fallon, Reference Ricklefs and Fallon2002; Waldenström et al. Reference Waldenström, Bensch, Kiboi, Hasselquist and Ottoson2002; Beadell et al. Reference Bensch, Pérez-Tris, Waldenström and Hellgren2004; Ishtiaq et al. Reference Ishtiaq, Gering, Rappole, Rahmani, Jhala, Dove, Milensky, Olson, Peirce and Fleischer2007), much of which was not evident from morphology alone (Hellgren et al. Reference Hellgren, Kristanauskiene, Valkiunas and Bensch2007). In addition, indications from the associations with corresponding nuclear sequences suggest that some cytochrome b lineages may represent reproductively isolated biological entities (Bensch et al. Reference Bensch, Pérez-Tris, Waldenström and Hellgren2004; Beadell et al. Reference Beadell, Covas, Gebhard, Ishtiaq, Melo, Schmidt, Perkins, Graves and Fleischer2009). Employing a molecular approach gives us the opportunity to gain a deeper understanding of the diversity of avian haematozoa present and, by resolving the history of association, to gain insight into the threat posed to the endemic avifauna by novel parasite introductions.
In this study, using mitochondrial DNA sequences (cytochrome b gene), we investigated geographical structuring of Plasmodium and Haemoproteus across avian hosts from the following islands or archipelagos in the Indian Ocean region (Fig. 1): Madagascar, Mascarenes (Mauritius, Reunion and Rodrigues), Comoros (Grand Comore, Moheli, Anjouan and Mayotte) and granitic Seychelles (of which the principal islands are Mahe, Silhoutte, La Digue and Praslin). Specifically, we aimed to characterize (a) haematozoan parasite diversity; (b) the phylogenetic relationships of haematozoan parasites found in these islands and (c) the evolutionary relationships between mainland and island parasite lineages.
MATERIALS AND METHODS
Avian sampling
A total of 150 avian blood samples from western Indian Ocean islands were collected during expeditions to Madagascar, the Mascarenes, Comoros and granitic Seychelles between July 1999 and February 2002 (Table 1 for species and sample size). All samples were taken non-destructively from mist-netted individuals and stored in Queen's lysis buffer (Seutin et al. Reference Seutin, White and Boag1991, Reference Seutin, Brawn, Ricklefs and Bermingham1993). In addition, 145 blood samples from mainland Africa were collected from 1991 to 2005 (see Appendix 1).
Table 1. Prevalence and molecular ID of haematozoan parasites in avian hosts sampled across islands of the western Indian Ocean

Molecular analysis
Host and parasite DNA was extracted from blood samples using the Qiagen DNeasy kits and protocols. Samples were screened for haematozoan infection with primers F2/R2 (91 bp) and 850F/1024R (167 bp; Beadell et al. Reference Beadell, Gering, Austin, Dumbacher, Peirce, Pratt, Atkinson and Fleischer2004) designed to amplify small fragments of cytochrome b gene and COIII gene, respectively. For those samples that were positive for parasites based on these tests, we attempted to amplify a larger fragment (256–533 bp) of cytochrome b using primers (F2/4292RW2-256 bp, FIFI/4292RW2-351 bp, F3/4292RW2-295 bp, or 3760/4292RW2-533 bp) designed from sequence that was relatively conserved between Haemoproteus and Plasmodium. Details of the polymerase chain reaction (PCR) were as reported by Beadell et al. (Reference Beadell, Gering, Austin, Dumbacher, Peirce, Pratt, Atkinson and Fleischer2004) and Ishtiaq et al. (Reference Ishtiaq, Beadell, Baker, Rahmani, Jhala and Fleischer2006). Negative and positive controls were included in all PCR reactions. In addition, to help ensure that any negative results from parasite screens were not attributable to poor quality of extractions, we amplified a small fragment (347 bp) of avian cytochrome b DNA using primers cytb-1/cytb2 (Kocher et al. Reference Kocher, Thomas, Meyer, Edwards, Pāābo, Villablanca and Wilson1989). These PCRs were successful in all cases.
We identified infections to genus by comparing the relationship of sequences obtained from this study to sequences in GenBank with known taxonomy (see Phylogenetic analysis below). In general, we sequenced the longest fragment that successfully amplified for any given sample. Samples were purified using Qiaquick (Qiagen) and sequenced bidirectionally on an ABI 3100 Sequencer (Applied Biosystems, Inc.). Sequences were edited and aligned using the program SEQUENCHER version 4.1 and are available through GenBank (Accession numbers JN661907–JN662002 in Appendix 1). In a few cases, we were only able to obtain sequence from the small fragments F2/R2 or 850F/1024R. In these cases, we identified the infection as Plasmodium or Haemoproteus, but we did not include these sequences in lineage-specific analyses. The adjusted Wald method was used to estimate 95% confidence intervals (CIs) for the estimates of parasite prevalence (Agresti and Coull, Reference Agresti and Coull1998).
Phylogenetic analysis
In order to estimate the relationships of western Indian Ocean parasite lineages to each other as well as to mainland lineages, we combined sequences obtained from western Indian Ocean samples with sequences obtained from additional PCR screening of mainland African hosts, as well as GenBank sequences derived from mainland African (Bensch et al. Reference Bensch, Stjernman, Hasselquist, Östman, Hansson, Westerdahl and Pinheiro2000; Waldenström et al. Reference Waldenström, Bensch, Kiboi, Hasselquist and Ottoson2002; Hellgren et al. Reference Hellgren, Kristanauskiene, Valkiunas and Bensch2007; Durrant et al. Reference Durrant, Reed, Jones, Dallimer, Cheke, McWilliam and Fleischer2007; Beadell et al. Reference Beadell, Covas, Gebhard, Ishtiaq, Melo, Schmidt, Perkins, Graves and Fleischer2009) and Asian hosts (Ishtiaq et al. Reference Ishtiaq, Gering, Rappole, Rahmani, Jhala, Dove, Milensky, Olson, Peirce and Fleischer2007) in a single phylogenetic analysis. In order to reduce the size of the tree for final analysis, we retained the single mainland lineage that was most closely related to each island-derived sequence, plus a random collection of other parasite lineages. We estimated the parasite phylogenetic relationships using samples for which we had at least 256 base pairs of cytochrome b sequence. We used MODELTEST version 3.6 (Posada and Crandall, Reference Posada and Crandall1998), to determine the most appropriate substitution model for our data. The hierarchical likelihood ratio test selected the general time reversible model (GTR+I+G). Phylogenetic reconstruction was implemented in BEAST version 1.4.7 (Drummond and Rambaut, Reference Drummond and Rambaut2007) using a MCMC (Markov Chain Monte Carlo) algorithm and assuming a relaxed clock model (Drummond et al. Reference Drummond, Ho, Phillips and Rambaut2006) of nucleotide substitution with a mean substitution rate of 1% per million years (and rates of evolution uncorrelated and log normal distribution among branches) and a Yule prior on branching. We conducted 2 runs for 20 million generations, each with sampling conducted every 1000 generations. Tracer (Rambaut and Drummond, Reference Rambaut and Drummond2003) was used to assess convergence, whether 2 chains were mixing, and whether the estimated sample (ESS) for each parameter was of sufficient size (ESS>200) to obtain robust parameter estimates. Four million generations were discarded as burn-in from each run, leaving a posterior distribution of 32 000 trees. We used 2 published sequences of mammalian Plasmodium parasites (see Perkins and Schall, Reference Perkins and Schall2002) as outgroups. The generic identity of the parasite lineages (Haemoproteus [H] vs Plasmodium [P]) was determined by the placement of cytochrome b sequences within a phylogenetic tree following Perkins and Schall (Reference Perkins and Schall2002), Beadell et al. (Reference Beadell, Gering, Austin, Dumbacher, Peirce, Pratt, Atkinson and Fleischer2004) and Ishtiaq et al. (Reference Ishtiaq, Beadell, Baker, Rahmani, Jhala and Fleischer2006, Reference Ishtiaq, Gering, Rappole, Rahmani, Jhala, Dove, Milensky, Olson, Peirce and Fleischer2007).
RESULTS
Parasite prevalence
Using a PCR-based approach, a total of 150 avian samples of 21 species was screened from the Indian Ocean region, of which 68 individuals (46 3%, 95% CI 38 3−54 3%) representing 16 species that tested positive for Plasmodium spp. or Haemoproteus spp. Of the samples testing positive for infection, 48 harboured Plasmodium, and 20 harboured Haemoproteus spp. infections. We detected Plasmodium spp. in samples from all 4 of the archipelagos; this parasite exhibited relatively high prevalence in Madagascar (46 4%; 95% CI 27 9−64 9%; n=28), the Comoros (50 8%; 95% CI 38 5−63 1%; n=63) and Mascarenes (40 9%; 95% CI 20 3−61 5%; n=22), compared to the Seychelles (8 1%; 95% CI 2 4−18 4%; n=37). We detected Haemoproteus spp. less frequently. While prevalence of Haemoproteus spp. was comparable to Plasmodium in Madagascar (39 3%; 95% CI 21 1−57 5%, n=28), prevalence was lower in the Comoros (14 3%; 95% CI 5 2−23 4%; n=63), Mascarenes (0%; n=22) and Seychelles (0%; n=37).
Parasite diversity
We detected a total of 16 distinct haematozoan lineages in the samples surveyed from the Indian Ocean region (Fig. 1). Madagascar and the Comoros exhibited the highest diversity of parasite lineages (10 and 9 lineages, respectively), while lineage diversity was lower in the Mascarenes (3 lineages) and the Seychelles (1 lineage). Lineage 3P accounted for 49% of all Plasmodium infections which were successfully sequenced. This lineage was detected in each of the 4 archipelagos and in 9 different host species. Lineage 8P was also relatively widely distributed, occurring in 3 of the 4 archipelagos, but only in Foudia madagascariensis.
Genetic relationships among parasite lineages
Of 145 mainland samples that were positive for haematozoa, 125 were successfully sequenced for parasite mitochondrial DNA. In total we identified 20 genetically distinct Plasmodium lineages (1P to 20P) and 21 Haemoproteus lineages (1H to 21H; Appendix 1). A comparison of these lineages with lineages detected in the Indian Ocean region, plus sequences available on GenBank, revealed only 4 cases of identical matches between island and mainland parasites. Among Plasmodium lineages, lineage 3P, which was widely distributed in the Indian Ocean, has also been detected in dozens of host species across the globe and, notably, on several remote island groups (Beadell et al. Reference Beadell, Ishtiaq, Covas, Melo, Warren, Atkinson, Bensch, Graves, Jhala, Peirce, Rahmani, Fonseca and Fleischer2006). Lineage 19P, which was found in Zosterops borbonicus from Mauritius matched GenBank sequence DQ368373, derived from Anthus trivialis (geographical origin unknown). Among lineages of Haemoproteus, lineage 3H retrieved from Hypsipetes madagascarensis matched identically to sequence retrieved from Pycnonotus nigricans in Botswana. Also, lineage 18H from Zosterops maderaspatanus and Z. m. kirki from Madagascar and Grand Comore, exhibited a sequence identical to parasites recovered from Tanzanian specimens of Zosterops senegalensis, Z. virens, and Z. poliogastra.
All parasite lineages fell into 2 distinct well-supported clades consistent with the genera Plasmodium and Haemoproteus (Fig. 2). Within each genus, lineages derived from the Indian Ocean islands did not form monophyletic groups, often exhibiting closer relationships to mainland-derived parasite lineages than other island lineages.

Fig. 2. Phylogenetic relationships of Plasmodium and Haemoproteus lineages based on maximum credibility clade using the Bayesian analysis. Shared lineages between mainland and island populations are in light grey squares and island shared lineages in dark grey squares. Parasite lineages from mainland Africa are labelled as BOS: Botswana; CAM: Cameroon; GAB: Gabon; KEN: Kenya; NIG: Nigeria; SEN: Senegal; SAF: South Africa; TAN: Tanzania. Lineages with prefix WA and WAH are from Beadell et al. (2009). Parasite lineages from islands are labelled as MAD=Madagascar; GDC=Grand Comore; MOH=Moheli; MAY=Mayotte; MAH=Mahe; SIL=Silhoutte; LDI=La Digue; PRA=Praslin; FRE=Fregate; REU=Reunion; MAU=Mauritius; ROD: Rodrigues. See Appendix 1 for details on host species.
DISCUSSION
In this study, which represents the first molecular survey of avian haematozoan parasites in the islands of the western Indian Ocean, we have identified multiple lineages of both Plasmodium spp. and Haemoproteus spp. in many of the endemic avian hosts in the region. Importantly, preliminary phylogenetic analysis of this parasite diversity suggests that these parasites do not constitute an endemic radiation and that at least certain lineages have likely colonized the islands relatively recently. We elaborate on these findings below.
Distribution of parasite lineages among islands
Haematozoan parasite community composition was heterogeneous across islands and archipelagos. While limited sampling may have amplified differences among islands, the observed geographical structuring of parasite lineages likely reflects the proximity of islands to mainland parasite sources, geographical area, avian host distribution, and the composition of vector communities on each island. Lineage composition on islands is also likely to be influenced by the extent of movement of vectors and hosts between islands as well as the host-specificity of parasites and their association with endemic or specialized vector species (e.g. Ishtiaq et al. Reference Ishtiaq, Guillaumot, Clegg, Phillimore, Black, Owens, Mundy and Sheldon2008). Although genetic differentiation between populations of several avian host families suggests that there is little or no movement of resident avifauna between islands (Nectariniidae, Warren et al. Reference Warren, Bermingham, Bowie, Prys-Jones and Thebaud2003; Zosteropidae, Warren et al. Reference Warren, Bermingham, Prys-Jones and Thebaud2006; Mila et al. Reference Mila, Warren, Heeb and Thébaud2010), human-mediated movements of birds has been extensive. In our survey, Madagascar and the Comoros exhibited the highest parasite lineage diversity, whereas the Seychelles and Mascarenes, more isolated archipelagos with smaller area and more limited avian diversity, exhibited lower parasite diversity. In the Indian Ocean region, we detected just 2 parasite lineages (LIN 3P, LIN 8P) that were found across 3 or more archipelagos. LIN 3P was the only lineage common across all 4 main island groups and occurred in 10 different host species. This parasite lineage (GRW4; AY099041, Bensch et al. Reference Bensch, Stjernman, Hasselquist, Östman, Hansson, Westerdahl and Pinheiro2000), which has colonized several other oceanic islands coincident with the global introduction of non-native birds over the last century (Beadell et al. Reference Beadell, Ishtiaq, Covas, Melo, Warren, Atkinson, Bensch, Graves, Jhala, Peirce, Rahmani, Fonseca and Fleischer2006), is likely to have followed the same route to the Indian Ocean region. This lineage shares a cytochrome b sequence identical to the strain of avian Plasmodium relictum (Beadell et al. Reference Beadell, Ishtiaq, Covas, Melo, Warren, Atkinson, Bensch, Graves, Jhala, Peirce, Rahmani, Fonseca and Fleischer2006) that decimated endemic bird populations in Hawaii (Warner Reference Warner1968; van Riper et al. Reference van Riper, van Riper, Goff and Laird1986), and therefore, a closer examination of its impact on the endemic avifauna of the Indian Ocean is warranted.
The other lineage with relatively wide distribution (LIN 8P), also appears to have extended its range via human-mediated host introductions. We recovered Plasmodium lineage 8P from Foudia madagascariensis in Madagascar, as well as Mayotte (Comoros) and Mauritius (Mascarenes). Foudia madagascariensis, which was the only species sampled across all 4 major archipelagos, is a Madagascar endemic that was introduced to many other islands in the Indian Ocean (B.H. Warren, Ph.D. thesis). Given the apparent specificity of LIN 8P, which was not recovered from any other host species, this lineage appears to have co-colonized Mayotte and Mauritius along with its introduced host.
Unfortunately, the distribution and vector competence of mosquitoes and other arthropod vectors have been little studied in the Indian Ocean region (but see Gerberg and Arnett, 1979) despite their likely influence on parasite distribution (Ishtiaq et al. Reference Ishtiaq, Guillaumot, Clegg, Phillimore, Black, Owens, Mundy and Sheldon2008). We did not detect Haemoproteus spp. in samples from the Seychelles or the Mascarenes, which might point to the absence of an appropriate vector. However, a microscopy-based survey by Lowery (Reference Lowery1971) detected Haemoproteus spp. in passerines from Aldabra (Seychelles) and Peirce et al. (Reference Peirce, Cheke and Cheke1977) reported Haemoproteus columbae only in samples of Columba livia in the Mascarenes. Our failure to detect Haemoproteus spp. in this survey, which did not include avian samples from Aldabra or C. livia from the Mascarenes, highlights the difficulty in fully capturing the structure of a diverse and complex parasite system. In many cases, the apparent absence of parasite lineages from particular hosts and islands may simply stem from limited sampling, temporary local extinctions, or infection intensities for certain lineages that frequently fall below levels of detection (e.g. Perkins, Reference Perkins2001; Fallon et al. Reference Fallon, Bermingham and Ricklefs2003). Also, differences in the timing of avian blood sampling could have resulted in discrepancies in the prevalence and distribution of certain parasite lineages; year-to-year temporal fluctuations in avian parasite prevalence and diversity have been documented (Wood et al. Reference Wood, Cosgrove, Wilkin, Knowles, Day and Sheldon2007), as have seasonal effects (Bensch and Ǻkesson, Reference Bensch and Ǻkesson2003; Cosgrove et al. Reference Cosgrove, Wood, Day and Sheldon2008). Whilst annual temporal (and spatial) variation in blood parasite prevalence is more pronounced in temperate faunas than in tropical ones (e.g. Schall and Marghoob, Reference Schall and Marghoob1995; Schall et al. Reference Schall, Pearson and Perkins2000; Freeman-Gallant et al. Reference Freeman-Gallant, O'Connor and Breuer2001; Fallon et al. Reference Fallon, Bermingham and Ricklefs2003), it should be noted that our sampling has been conducted over an extended time-period (1999–2002) and across a diverse range of islands with varying climatic regimes. Therefore temporal effects could also play a role in the observed heterogeneity of prevalence and diversity across islands.
Comparison between mainland and island parasite lineages
Sparse sampling of parasite lineages from continental Africa prevents us from drawing firm conclusions regarding the geographical and evolutionary origins of parasites in the Indian Ocean avifauna. In particular, we cannot be sure that we have recovered the continental lineage that is most closely related to any particular island lineage or that the apparent geographical or host distribution of continental lineages is representative. Nonetheless, 2 observations stand out. First, based on our phylogenetic analysis, the western Indian Ocean parasite lineages are polyphyletic with respect to mainland African lineages, suggesting that the Indian Ocean parasite community reflects multiple independent colonizations as opposed to a single endemic radiation of parasites. Second, the low divergence between several island and mainland parasites suggests that these parasite lineages may have colonized the islands relatively recently. We identified several Indian Ocean parasite lineages that were identical (LIN 3P, LIN 19P, LIN 3H LIN 18H) or nearly identical (genetic divergence 0 1–0 4%; LIN 8P, LIN 20P and LIN 8H, LIN 15H) to continental lineages across the segment of cytochrome b sequenced (minimum of 256 bp). Assuming that parasite divergence at cytochrome b occurs at approximately 1 2% per million years (Ricklefs and Outlaw, 2010), then identical sequences might be retrieved from lineages that have been diverging for anywhere between 0 and 325 000 years, the time expected to pass prior to a single mutation arising to distinguish 2 sequences of 256 bp. This time of divergence is low compared to the maximum estimated dates of island colonization for the avian host genera Nectarinia, Hypsipetes, Zosterops and Foudia, which are in the order of 1–3 million years before the present (Warren et al. Reference Warren, Bermingham, Bowie, Prys-Jones and Thebaud2003, Reference Warren, Bermingham, Prys-Jones and Thebaud2005, Reference Warren, Bermingham, Prys-Jones and Thebaud2006). Thus, on the time-scale of mutation in the cytochrome b gene, at least some parasite lineages have been exchanged between continental Africa and neighbouring islands after island hosts became isolated.
Parasite exchange may have been facilitated by rare migrants or by human-mediated introductions of species such as common waxbills (Estrilda estrild), house sparrows (Passer domesticus), common mynas (Acridotheres tristis) and red-whiskered bulbuls (Pycnonotus cafer), which occur throughout the archipelagos. An interesting exception to this model, however, might be Haemoproteus lineage LIN 18H, which was isolated exclusively from Zosterops spp. in both Tanzania and the Indian Ocean. Given the apparent host specificity of this lineage and the absence of any introduced species of Zosterops in the Indian Ocean, this parasite may have instead arrived in the islands via an infected vector or, alternatively, may have been present in the first Zosterops colonist. If the latter is true, then the lack of divergence in LIN 18H will have to be reconciled with the relatively long time of divergence estimated for Zosterops spp. in the Indian Ocean (Warren et al. Reference Warren, Bermingham, Prys-Jones and Thebaud2006). To further clarify the origins of Indian Ocean parasites, future work should be directed towards characterizing the molecular relationships between mainland African parasites and parasites deemed to be endemic to the Indian Ocean region based on morphology (Savage et al. Reference Savage, Robert, Goodman, Raharimanga, Raherilalao, Andrianarimisa, Ariey and Greiner2009).
CONCLUSIONS
Our initial molecular survey of vector-mediated parasites in the western Indian Ocean provided the following insights into the distribution and diversity of parasite lineages: (a) parasite communities exhibited heterogeneity in prevalence and composition across the islands, (b) parasite communities were diverse, consistent with the findings of microscopy-based surveys, (c) the phylogenetic relationships among parasite lineages appear to reflect multiple independent colonizations of the western Indian Ocean region, and (d) in many cases, these colonizations appear to have occurred relatively recently based on the sharing of parasite lineages between mainland Africa and Indian Ocean Islands. Given the relatively small, geographically restricted and sometimes threatened populations of avian hosts present in the Indian Ocean region, assessing the ecological impact of haematozoan parasites on the endemic island avifauna should be a priority.
FINANCIAL SUPPORT
This study was funded by the National Institutes of Health grant no. 1R01GM063258. We thank C. McIntosh for facilitating laboratory analyses. We are grateful to S. Anli, T. Ghestemme, C. Moussa, Iboura, R. Nichols, F. Ratrimomanarivo and I. Saïd for help in the field in the Indian Ocean. We also thank the following Indian Ocean organizations for granting permission and logistical support for the fieldwork: CNDRS, CNDRS d'Anjouan, Conservation de la Biodiversité Moheli, DAF-SEF, MEF Madagascar, MHN La Réunion, MICET, MNPCS, MWF, Nature Seychelles, SBS, SDOE, SEOR and SIF.
APPENDIX 1
List of haematozoan lineages with GenBank Accession numbers. The taxonomy follows Sibley and Monroe (1996)




