


INTRODUCTION
The rickettsial bacterium Ehrlichia ruminantium is the causative pathogen of heartwater in ruminants. The organism is transmitted by ticks of the genus Amblyomma and is responsible for considerable livestock losses in endemic countries (Mukhebi et al. Reference Mukhebi, Chamboko, O'Callaghan, Peter, Kruska, Medley, Mahan and Perry1999), and is a potential emerging zoonosis as suspected human cases have been reported (Allsopp et al. Reference Allsopp, Louw and Meyer2005; Louw et al. Reference Louw, Allsopp and Meyer2005). Heartwater is prevalent in nearly all of sub-Saharan Africa and also on some islands in the Caribbean, where it poses the threat of spreading to the American mainland (Burridge et al. Reference Burridge, Simmons, Peter and Mahan2002). There is only 1 commercially available vaccine against heartwater, which utilizes Ball 3 E. ruminantium strain in an infection and treatment strategy. This has been used extensively over many years in South Africa (Oberem and Bezuidenhout, Reference Oberem and Bezuidenhout1987), but it provides relatively limited cross-protection against other strains (Du Plessis et al. Reference Du Plessis, van Gas, Olivier and Bezuidenhout1989). Although several types of experimental vaccines, including inactivated (Martinez et al. Reference Martinez, Maillard, Coisne, Sheikboudou and Bensaid1994; Mahan et al. Reference Mahan, Andrew, Tebele, Burridge and Barbet1995, Reference Mahan, Smith, Kumbula, Burridge and Barbet2001), attenuated (Jongejan, Reference Jongejan1991; Zweygarth et al. Reference Zweygarth, Josemans, van Strijp, Lopez-Rebollar, van Kleef and Allsopp2005; Faburay et al. Reference Faburay, Geysen, Ceesay, Marcelino, Alves, Taoufik, Postigo, Bell-Sakyi and Jongejan2007a), recombinant (Pretorius et al. Reference Pretorius, Liebenberg, Louw, Collins and Allsopp2010), and DNA vaccines (Pretorius et al. Reference Pretorius, van Kleef, Collins, Tshikudo, Louw, Faber, van Strijp and Allsopp2008), have been developed to control the disease, these have demonstrated limited efficacy in field trials (Mahan et al. Reference Mahan, Smith, Kumbula, Burridge and Barbet2001; Faburay et al. Reference Faburay, Geysen, Ceesay, Marcelino, Alves, Taoufik, Postigo, Bell-Sakyi and Jongejan2007a). It has been experimentally shown that effective cross-protection is not necessarily defined by the geographical origins of the isolates – it is possible for there to be a complete lack of cross-protection between isolates originating from a small geographical area and conversely, partial or complete cross-protection between isolates even from widely separated geographical areas (van Winkelhoff and Uilenberg, Reference van Winkelhoff and Uilenberg1981; Uilenberg et al. Reference Uilenberg, Zivković, Dwinger, ter Huurne and Perié1983; Jongejan et al. Reference Jongejan, Uilenberg, Franssen, Gueye and Nieuwenhuijs1988; Du Plessis et al. Reference Du Plessis, van Gas, Olivier and Bezuidenhout1989). One obvious hypothesis is that vaccine efficacy is highly dependent upon the E. ruminantium genotypes circulating in the particular region (Faburay et al. Reference Faburay, Geysen, Munstermann, Taoufik, Postigo and Jongejan2007b; Adakal et al. Reference Adakal, Stachurski, Konkobo, Zoungrana, Meyer, Pinarello, Aprelon, Marcelino, Alves, Martinez, Lefrancois and Vachiéry2010b). Thus, the proper characterization of field isolates of E. ruminantium is a necessary component for an effective vaccine strategy.
Molecular genotyping will make it feasible to explore correlations between E. ruminantium genotypes and vaccine efficacy, and to provide information on the genetic populations in the area prior to the appropriate formulation of the vaccine. Several methods have been developed to genotype E. ruminantium. The map1 gene, encoding for a major antigenic surface protein, showed high levels of sequence polymorphism between strains (Reddy et al. Reference Reddy, Sulsona, Harrison, Mahan, Burridge and Barbet1996; Raliniaina et al. Reference Raliniaina, Meyer, Pinarello, Sheikboudou, Emboulé, Kandassamy, Adakal, Stachurski, Martinez, Lefrançois and Vachiéry2010) and was successfully used to genotype field isolates by sequence typing (Martinez et al. Reference Martinez, Vachiéry, Stachurski, Kandassamy, Raliniaina, Aprelon and Gueye2004) and PCR-restriction fragment length polymorphism (PCR-RFLP) analysis (Martinez et al. Reference Martinez, Vachiéry, Stachurski, Kandassamy, Raliniaina, Aprelon and Gueye2004; Faburay et al. Reference Faburay, Jongejan, Taoufik, Ceesay and Geysen2008). However, typing based on this gene did not show any correlation with virulence, vaccine efficacy, or geographical origins (Allsopp et al. Reference Allsopp, Dorfling, Maillard, Bensaid, Haydon, van Heerden and Allsopp2001; Martinez et al. Reference Martinez, Vachiéry, Stachurski, Kandassamy, Raliniaina, Aprelon and Gueye2004). Recently, multilocus sequence typing (MLST) analysis was developed based on 8 housekeeping genes (Adakal et al. Reference Adakal, Meyer, Carasco-Lacombe, Pinarello, Allègre, Huber, Stachurski, Morand, Martinez, Lefrançois, Vachiery and Frutos2009). Although this assay could discriminate between even closely related isolates circulating in the same area, it is very expensive and labour intensive to perform with a large number of samples. Moreover, our recent study showed that this method failed to discriminate between 2 closely related strains, Kerr Seringe and Sankat 430 (Nakao et al. Reference Nakao, Magona, Zhou, Jongejan and Sugimoto2011). There is, consequently, still a need for a novel genotyping method that combines high discriminatory power with cost-effectiveness.
Multiple-locus variable-number tandem-repeat (VNTR) analysis (MLVA) is a prominent typing tool which has been successfully applied to genotyping of a variety of bacterial species (Lindstedt, Reference Lindstedt2005; van Belkum, Reference van Belkum2007). This method is based on the detection of the number of repeats at VNTR loci spread throughout the bacterial genome. Variability of the number of repeats is used to produce allele profiles for each isolate. Analysis with capillary electrophoresis combined with multiplex PCR assays with sets of fluorescently labelled primers allows rapid, accurate, and cost-effective evaluation of VNTRs.
The aim of this study was to identify potential VNTRs for use in a MLVA genotyping scheme using 17 E. ruminantium reference strains from geographically diverse origins. We further developed multiplex PCR assays targeting 8 VNTRs with fluorescently tagged primers and a rapid fragment separation using an automated capillary electrophoresis. This high-throughput typing assay was then applied to E. ruminantium in Amblyomma variegatum collected from indigenous cattle in 6 different districts of Uganda where heartwater is endemic. MLVA revealed that there were different genotypes circulating in the study area, and the discriminatory power of MLVA was greater than that of map1 PCR-restriction fragment length polymorphism analysis.
MATERIALS AND METHODS
Ehrlichia ruminantium reference strains
Details of E. ruminantium reference strains used in this study are presented in Table 1. All were cultured in bovine aorta endothelium cells as described previously (Jongejan, Reference Jongejan1991) and subjected to DNA extraction at passages 2–3 after initiation of cultures from frozen blood or infected-cell stabilates. Attenuated Senegal (Jongejan, Reference Jongejan1991) at passage 14 was included to determine the stability of each locus.
Table 1. Ehrlichia ruminantium strains used in this study

a The numbers following the letters A, T, and C represent the numbers of passages in animals, ticks, and cell cultures, respectively. When the passage history prior to the storage in Utrecht Centre for Tick-borne Diseases (UCTD), the Netherlands is unknown, only passage history in UCTD is shown in parentheses.
b NR, not recorded in the reference.
c NA, not applicable.
Tick samples
From December 2008 to January 2009, adult A. variegatum were collected from indigenous cattle in 6 districts in Uganda (see Supplementary Fig. 1; online version only): Amuria (33.38°W, 02.01°N), Butaleja (33.56°W, 00.55°N), Dokolo (33.10°W, 01.55°N), Pallisa (33.42°W, 01.10°N), Soroti (33.36°W, 01.43°N), and Tororo (34.11°W, 00.41°N). Live ticks were stored in sealed plastic bags containing silica gel until used for DNA extraction. In total, 735 ticks, including 120 samples used in a previous study (Nakao et al. Reference Nakao, Stromdahl, Magona, Faburay, Namangala, Malele, Inoue, Geysen, Kajino, Jongejan and Sugimoto2010), were tested for E. ruminantium infection by pCS20 PCR (Van Heerden et al. Reference van Heerden, Steyn, Allsopp, Zweygarth, Josemans and Allsopp2004).
DNA extraction
DNA from rickettsia-infected cell cultures was extracted using Nucleospin Tissue kits (Macherey-Nagel, Duren, Germany). Whole dead A. variegatum ticks, dessicated by storage with silica gel, were washed with 70% ethanol and rinsed twice with distilled water. Ticks were then homogenized by Micro Smash MS-100R (TOMY, Tokyo, Japan) for 2 min at 2500 rpm, followed by DNA extraction with DNAzol (Invitrogen, Carlsbad, CA, USA). All procedures were carried out as described by the manufacturers.
Identification and selection of VNTR loci in Ehrlichia ruminantium
The genomes of 3 E. ruminantium strains, Welgevonden (Erwo) (GenBank Accession no. CR767821) (Collins et al. Reference Collins, Liebenberg, de Villiers, Brayton, Louw, Pretorius, Faber, van Heerden, Josemans, van Kleef, Steyn, van Strijp, Zweygarth, Jongejan, Maillard, Berthier, Botha, Joubert, Corton, Thomson, Allsopp and Allsopp2005), Welgevonden (Erwe) (GenBank Accession no. CR925678), and Gardel (Erga) strains (GenBank accession no. CR925677) (Frutos et al. Reference Frutos, Viari, Ferraz, Morgat, Eychenié, Kandassamy, Chantal, Bensaid, Coissac, Vachiery, Demaille and Martinez2006), were explored for potential VNTR loci using Tandem Repeat Finder program version 4.04 (Benson, Reference Benson1999). Subsequently, loci were selected based on the following criteria: (i) repeat size should be less than 100 bp; (ii) the position in the genome should be conserved between 3 strains; (iii) repeat numbers should be identical between Erwo and Erwe; (iv) the sequences flanking the tandem-repeat region should be conserved between 3 strains and have adequate GC% to design primers; (v) PCR amplicon size should be less than 600 bp for optimal use in capillary electrophoresis analysis. When the loci met the criteria, PCR primers flanking the tandem-repeat regions were designed using Primer3Plus software (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi). Each candidate tandem-repeat locus was screened by PCR amplification using 17 reference strains from geographically diverse origins, including 1 attenuated strain. Initially, the variability of each tandem-repeat locus was assessed by gel electrophoresis on a 1·2% agarose gel. To confirm that length polymorphisms were due to variation in copy number, the PCR products were purified with ExoSAP-IT (USB Corporation, Cleveland, OH, USA) and sequenced using the BigDye Terminator version 3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) and an ABI Prism 3130x genetic analyzer (Applied Biosystems) according to the manufacturers' instructions. The 5′ and 3′ flanking regions of each tandem repeat were confirmed by comparison with the available E. ruminantium genomic sequence, and the tandem-repeat sequences were counted manually to determine the repeat copy number at each locus for each E. ruminantium strain according to the following formula: Repeat copy number= [Amplicon size (bp) – Flanking regions (bp)]/Repeat size (bp).
Multiplex PCR assays
In total, 8 VNTR loci were selected for MLVA and amplified in 2 separate multiplex amplification reactions named multiplex 1 and 2. Multiplex 1 consisted of ERTR12-1, ERTR24-1, ERTR10-1, and ERTR27-2, while multiplex 2 consisted of ERTR06-2, ERTR24-2, ERTR12-2, and ERTR22-1. The forward primers of each primer pair were fluorescently labelled with either 6-FAM, VIC, NED or PET at the 5′ end, while the reverse primers were synthesized with pigtail at the 3′ end (Applied Biosystems). The primer sequences with corresponding fluorochromes and final concentration in the reaction mixture are listed in Table 2. The PCRs were performed using Multiplex PCR Assay Kit (TaKaRa Bio, Otsu, Japan). All reactions were conducted in a 25-μl reaction mixture containing 12·5 μl of Multiplex PCR Mix 2, 0·125 μl of Multiplex PCR Mix 1, 1·0 μl of primer mix, 1·0 μl of template DNA, and distilled water. Cycling conditions were as follows: an initial denaturation step at 94°C for 60 sec; 40 cycles of denaturation at 94°C for 30 sec, annealing at 58°C for 90 sec, and elongation at 72°C for 90 sec. The amplifications were terminated after a final elongation step at 72°C for 10 min.
Table 2. Primers used for multiplex PCR assays in MLVA typing with capillary electrophoresis

a Fluorescent labels shown in bold.
b PCR amplicon size in Welgevonden strain.
Capillary electrophoresis of fluorescently labelled PCR products
Prior to capillary electrophoresis, PCR products from the 2 multiplex reactions were diluted 1:100 in distilled water. One μl of the diluted samples was mixed with 10 μl of Hi-Di formamide (Applied Biosystems) and 0·5 μl of GeneScan 600 LIZ Size Standard (Applied Biosystems). After heat denaturation at 95°C for 5 min and rapid cooling on ice, the fragments were separated by capillary electrophoresis using the ABI Prism 3130x genetic analyzer. The fragment analysis was performed using Gene Mapper software version 4.0 (Applied Biosystems). The repeat copy number at each locus was determined as a whole number according to the following formula: Repeat copy number=[Flagment size (bp) – Flanking regions (bp) – Pigtail (7 bp)]/Repeat size (bp).
Data analysis
The data with the calculated number of repeats were imported into BioNumerics software version 6.5 (Applied Maths, Saint-Martens-Latem, Belgium) and Simpson's index of diversity was calculated for each VNTR. Analysis of genotypes used Clustering Calculator (http://www2.biology.ualberta.ca/jbrzusto/cluster.php) to generate a Phylip Drawtree string (unweighted arithmetic average clustering method, and Jaccard's similarity coefficient), which was converted into a dendrogram by Treeview (http://taxonomy.zoology.gla.ac.uk/rod/treeview.html) (Page, Reference Page1996). Clustering Calculator generated the bootstrap values for dendrograms, using 100 iterations. Principal Component Analysis (PCA) of the genotypes was performed using the GenAlEx program (Peakall and Smouse, Reference Peakall and Smouse2006) following determination of genetic distance with data standardisation. A minimum-spanning tree (MST) was generated using BioNumerics software for visualization of the relationship between MLVA profiles in a single compact image.
PCR-RFLP analysis
PCR-RFLP analysis was conducted on field samples that were positive for E. ruminantium by pCS20 PCR. PCR amplification was conducted using nested map1 PCR, followed by digestion of the PCR products with restriction enzyme Alu I as described previously (Faburay et al. Reference Faburay, Jongejan, Taoufik, Ceesay and Geysen2008). The digested fragments were separated on a 3·5% agarose gel (Agarose Zebra; BioTools, Tokyo, Japan) at 50 V for 120 min in 0.5x TBE buffer. A 100 bp ladder was included to determine the fragment size. The gel was subjected to GelRed TM staining (Biotium, Hayward, CA, USA) and visualized under UV light.
RESULTS
Identification of VNTR loci in Ehrlichia ruminantium
The Tandem Repeat Finder software identified 201, 192, and 179 repeat loci within E. ruminantium Erwo, Erwe, and Erga strains, respectively. Initially, 30 repeat loci were selected according to the above-mentioned criteria and primers flanking each tandem-repeat region were designed (Supplementary Table S1; online version only). Subsequently, PCR amplification of each locus was tested using a panel of 17 strains from geographically diverse origins, including 1 attenuated strain (Table 1). When single bands in all tested strains were obtained in gel electrophoresis of PCR products, sequencing analysis was conducted to detect the variability of repeat numbers. For 6 VNTRs, specific primers failed to amplify the DNA satisfactorily (Table 3). Sequencing analysis revealed that 8 VNTRs were monomorphic and that 4 VNTRs showed polymorphisms, which were not only related to variations in repeat number but also to insertions or deletions in the flanking regions. The remaining 12 loci had more than 1 allele among the tested strains and were named as follows: ERTR+repeat size+differentiation number (ER stands for E. ruminantium). The length of repeat ranged from 6 bp of ERTR06-01 and ERTR06-02 to 90 bp of ERTR90-01. Two of 12 VNTRs were located in intergenic regions, while 10 were identified in open reading frames (ORFs). ERTR06-01 and ERTR06-02 were respectively localized in the virD4 and virB10 genes, which encode the type IV secretion system proteins. The other 8 VNTRs were located in ORFs coding for hypothetical proteins. A schematic map of the genomic positions of 12 VNTR loci for the E. ruminantium Welgevonden (Erwo) strain is shown in Fig. 1.

Fig. 1. Schematic map of 12 VNTR loci on Welgevonden (Erwo) strain genome. Each bar represents the VNTR locus and the numbers inside the circle indicate the positions within the genome of the Welgevonden (Erwo) strain.
Table 3. Results of initial screening of potential VNTRs by PCR and sequencing

a Location on the genome sequence of Welgevonden (Erwo) strain.
b Copy number in Welgevonden strain.
c PCR amplification was conducted using 17 E. ruminantium reference strains.
d NA, not applicable.
e Yes*, repeat sequence is absent or not conserved in all strains.
f TR which has more than 2 alleles.
Characteristics of VNTRs in 16 Ehrlichia ruminantium reference strains
Allelic profiles of 12 VNTRs in 16 E. ruminantium reference strains are shown in Table 4. The number of alleles identified in the tested strains ranged from 2 to 14, and the Simpson's diversity indices (DIs) of 12 VNTRs differed considerably. The lowest DI (12·5) was found for ERTR21-1, ERTR27-1, and ERTR90-1, and the highest (98·3) was for ERTR12-1. None of the 12 VNTRs were altered between original and attenuated Senegal strain (data not shown), indicating that the VNTR loci are stable and do not change, at least under prolonged maintenance in in vitro culture.
Table 4. Allelic profiles of 12 VNTRs in Ehrlichia ruminantium reference strains

a DI, Simpson's diversity index.
Cluster analysis using 12 VNTR profiles
Tandem-repeat loci that generated more than 1 allele were selected for a cluster analysis of 16 E. ruminantium reference strains. The clustering analysis revealed the existence of 3 major clusters (clusters A, B, and C, Fig. 2). Cluster A consists of 3 strains from South Africa and 1 each from Burkina Faso, São Tomé and Principe, and Sudan. Cluster B consists of strains from diverse origins including Kenya, Nigeria, South Africa, Zambia, and the Caribbean. There was moderate support for the separation of these clusters (bootstrap value of 31), but very strong support for the distinction between clusters C and A/B (bootstrap=100). Cluster C consists of 1 strain from Zimbabwe and 4 strains from West Africa (Senegal, The Gambia, and Ghana). The latter 4 strains were able to be further divided into a subgroup (C1) by clustering analysis (bootstrap=44). PCA highlights the split between group C and groups A/B, with principal coordinate 1 accounting for 31% of the variation observed in the whole dataset (Fig. 3). From the clustering analysis and PCA, there was no correlation between clusters and geographical origins observed, apart from cluster C1 all originating from West Africa.

Fig. 2. Dendrogram of 12 VNTRs from Ehrlichia ruminantium reference strains. Bootstrap values as calculated from 100 iterations are shown at major nodes only.

Fig. 3. Principle Component Analysis of Ehrlichia ruminantium reference strains. Coordinate 1 accounts for 31% of the variation observed in the dataset and separates cluster C from clusters A and B. Symbols represent geographical origin: diamond=Southern Africa, square=West Africa, triangle=East Africa, and circle=Caribbean.
Selection of VNTRs and primer combinations for multiplex PCR assays
We then developed 2 multiplex PCR systems for rapid, simple, and accurate detection of VNTR alleles by capillary electrophoresis. From the 12 VNTRs, 8 with higher DI were selected, for which fluorescently labelled primers were designed as indicated in Table 2. ERTR06-1 was not included in the assay since there was a deletion in the 5′ flanking region of the repeat locus, making it difficult to identify variations in repeat number by fragment sizes. All of the combinations and primer concentrations were optimized using DNA from the Welgevonden strain. The GeneScan 600 LIZ Size Standard generated peaks from 20 to 600 bp, which covered all the fragment sizes resulting from multiplex PCR reactions. Figure 4 shows representative electrophoresis of 2 multiplex PCR assays on Welgevonden strain.

Fig. 4. Representative electropherograms after capillary electrophoresis separation of the fragments amplified by 2 multiplex PCR assays. Upper and lower panels represent multiplex 1 and 2 in Welgevonden strain, respectively. The orange peaks are the LIZ-labelled internal size standards. ERTR27-2 and ERER06-2 were labelled with 6-FAM (blue peaks), ERTR24-1 and ERER12-2 were labelled with VIC (green peaks), ERTR10-1 and ERER24-2 were labelled with NED (black peaks), and ERTR12-1 and ERER22-1 were labelled with PET (red peaks).
Detection of Ehrlichia ruminantium in Amblyomma variegatum from Uganda by pCS20 PCR
Out of 735 A. variegatum, 38 tested positive for E. ruminantium by pCS20 PCR (overall positive rate of 5·2%). The highest and lowest positive rates were 6·8% in Tororo and 1·4% in Dokolo, respectively (Table 5). We did not record the individual host that each tick was sampled from, and therefore the data are not available to discuss whether the E. ruminantium-positive ticks were associated with individual or multiple animals from each herd of cattle at the sampling site.
Table 5. Summary of pCS20 PCR detection, map1 PCR-RFLP and MLVA typing of Ehrlichia ruminantium in Amblyomma variegatum collected in six districts in Uganda
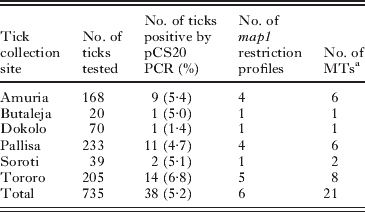
a MTs, MLVA types.
PCR-RFLP analysis
The map1 nested PCR amplified specific amplicons from all E. ruminantium-positive A. variegatum. Subsequent digestion with Alu I resulted in 6 different restriction profiles within 38 tick samples (Fig. 5 and Table 5). Profile I showed highest overall frequency of 39% (n=15), followed by profile IV (n=9), profile III (n=8), and profile V (n=4). Profiles II and VI were detected only once (Table 6). The restriction profiles did not show any geographical clustering among different samples.

Fig. 5. Restriction profiles of map1 gene digested with Alu I. Lanes: 1, A004 (restriction profile I); 2, A022 (profile II); 3, A034 (profile III); 4, A054 (profile IV); 5, P016 (profile V); 6, T009 (profile VI); M, 100 bp marker.
Table 6. Allelic profiles of MLVA and map1 restriction profiles of Ehrlichia ruminantium in Amblyomma variegatum

a MT, MLVA type.
b DI, Simpsons diversity index.
MLVA
A set of 8 VNTRs was used to type 38 E. ruminantium-positive ticks collected in Uganda. All VNTRs were successfully amplified from all E. ruminantium-positive ticks (Table 6). The number of alleles identified within the 38 ticks ranged from 1 (ERTR12-2, ERTR22-1 and ERTR24-1) to 11 (ERTR12-1) and the DIs ranged from 0·0 (ERTR12-2, ERTR22-1 and ERTR24-1) to 82·3 (ERTR27-2). In total, 21 MLVA types (MTs) were recovered from 38 tick samples. Of these types, MT-12 was predominant and found in 7 ticks, followed by MT-2 in 6 ticks. In contrast, 14 MTs were recovered only once from a single tick. Tick T104 yielded 2 alleles at locus ERTR24-2 indicating infection with more than one E. ruminantium genotype, while in the remaining 37 ticks 1 allele was found at each locus, suggesting they were infected with only a single E. ruminantium genotype. All MTs of samples from Uganda were distinct from those of the 16 E. ruminantium reference strains. MST analysis conducted on MLVA profiles of reference strains and field samples from Uganda identified 4 main groups (named A, B, C, and D) (Fig. 6). Group A was the largest group consisting of 26 samples (68·4% of all samples from Uganda). Groups B, C, and D were composed of 8, 2, and 2 samples, respectively. One sample, S013, was not included in any groups. None of the samples from Uganda clustered with reference strains from other countries. There was no correlation observed between groups and collection sites. The comparison between map1 PCR-RFLP types and MST groups showed that map1 PCR-RFLP types, except for profile IV, were well correlated with MST groups (Table 7).

Fig. 6. Minimum spanning tree of MLVA profiles of 16 Ehrlichia ruminantium reference strains and 38 E. ruminantium-infected A. variegatum from Uganda. Each circle in the tree represents a different MLVA type, and the type number is indicated by the number in the circle. The circle colours indicate the regions where the ticks were collected. Circle size is proportional to the numbers of samples belonging to an MLVA type. Numbers between the circles represent the summed tandem-repeat difference between MLVA types. Two or more MLVA types differing at 1 locus are regarded as a group and are marked in colour.
Table 7. Comparison between map1 restriction profiles and MST groups recovered from field samples from Uganda
DISCUSSION
The availability of complete genome sequences of different strains of E. ruminantium has given rise to the possibility of using genetic markers at the whole genome scale for use in reliable genotyping. One of the genomic features of this bacterium is the presence of a large number of tandem repeat sequences throughout the genome (Collins et al. Reference Collins, Liebenberg, de Villiers, Brayton, Louw, Pretorius, Faber, van Heerden, Josemans, van Kleef, Steyn, van Strijp, Zweygarth, Jongejan, Maillard, Berthier, Botha, Joubert, Corton, Thomson, Allsopp and Allsopp2005; Frutos et al. Reference Frutos, Viari, Ferraz, Morgat, Eychenié, Kandassamy, Chantal, Bensaid, Coissac, Vachiery, Demaille and Martinez2006). In this study, such repetitive units have been used as informative markers in MLVA genotyping, which has been shown in other bacteria to have high discriminatory power as well as reproducibility between different laboratories (Lindstedt, Reference Lindstedt2005; van Belkum, Reference van Belkum2007). To our knowledge this is the first report of molecular typing of E. ruminantium based on MLVA profiles.
Stability of the MLVA markers is a prerequisite for reliable molecular typing. Since tandem repeat sequences in the E. ruminantium genome are reported to be largely responsible for the ongoing process of its genomic plasticity (Collins et al. Reference Collins, Liebenberg, de Villiers, Brayton, Louw, Pretorius, Faber, van Heerden, Josemans, van Kleef, Steyn, van Strijp, Zweygarth, Jongejan, Maillard, Berthier, Botha, Joubert, Corton, Thomson, Allsopp and Allsopp2005; Frutos et al. Reference Frutos, Viari, Ferraz, Morgat, Eychenié, Kandassamy, Chantal, Bensaid, Coissac, Vachiery, Demaille and Martinez2006), some are too variable to use for a fingerprinting method and thus the selection of stable VNTRs was essential. In this study, therefore, we excluded VNTRs with repeat numbers differing between Erwo and Erwe, which originated from the same source but were maintained at different laboratories for over 18 years. In addition, the conservation of repeat numbers between the original Senegal isolate and its attenuated vaccine strain, generated by continuous in vitro passage, provided a degree of confidence in the stabilities of the VNTRs identified in this study.
To increase throughput, we have developed a multiplex PCR approach suitable for rapid and simultaneous amplification of multiple targets. In addition, the use of the internal size standard combined with capillary electrophoresis separation enabled more precise fragment sizing compared to standard gel electrophoresis. Likewise, MLVA with high throughput capacity has clear advantages over sequencing-based typing in that it is simple, quick and relatively cheap to perform, which are critical factors for application to epidemiological studies where a large number of samples may be required for analysis. MLVA described in this study was able to discriminate between Kerr Seringe and Sankat 430, which was not achieved by MLST analysis in our recent study (Nakao et al. Reference Nakao, Magona, Zhou, Jongejan and Sugimoto2011). Moreover, while MLST failed to discriminate between A004, A006, and T009, and between D002 and P006 (Nakao et al. Reference Nakao, Magona, Zhou, Jongejan and Sugimoto2011), MLVA was able to discriminate between A004 and A006, between A004 and T009, and between D002 and P006. Although further comparison of these two genotyping methods using a larger number of samples is required, from this dataset a preliminary conclusion is that MLVA has higher discriminatory power than MLST.
One tick sample, T104, yielded multiple fragments with a marker ERTR24-2. Further cloning and sequencing analysis of PCR products confirmed that they were not due to nonspecific PCR amplifications but more likely due to multiple infections of different genotypes of E. ruminantium in a single tick (data not shown). Compared to the previous methods such as PCR-RFLP (Faburay et al. Reference Faburay, Jongejan, Taoufik, Ceesay and Geysen2008) and sequencing of cloned PCR products (Vachiéry et al. Reference Vachiéry, Raliniaina, Stachurski, Adakal, Molia, Lefrançois and Martinez2006; Barbet et al. Reference Barbet, Byrom and Mahan2009), MLVA is likely to be able to detect multiple infections more easily and quickly. Since this could be another advantage of MLVA in the analysis of field samples, where the presence of mixed population in a single animal or tick has been previously reported (Faburay et al. Reference Faburay, Jongejan, Taoufik, Ceesay and Geysen2008; Barbet et al. Reference Barbet, Byrom and Mahan2009), further evaluation using animals or ticks that have been experimentally infected with 2 different E. ruminantium genotypes is required.
As shown by Frutos et al. (Reference Frutos, Viari, Ferraz, Morgat, Eychenié, Kandassamy, Chantal, Bensaid, Coissac, Vachiery, Demaille and Martinez2006), the size of tandem repeat units is distributed bimodally in E. ruminantium with small repeats of approximately 12 bp and long-period repeats ranging from 100 to 300 bp being the most frequent. The former, which were mainly explored for screening in this study, comprise only 15% of the total number of tandem repeats (Frutos et al. Reference Frutos, Viari, Ferraz, Morgat, Eychenié, Kandassamy, Chantal, Bensaid, Coissac, Vachiery, Demaille and Martinez2006), indicating that there are still a large number of VNTRs that may be likely candidates for further informative markers. Another possible approach to refine our method would be a combination with other genotyping methods such as MLST- and SNP-based genotyping as reported for other bacteria (Octavia and Lan, Reference Octavia and Lan2009; Marsh et al. Reference Marsh, O'Leary, Shutt, Sambol, Johnson, Gerding and Harrison2010; Valdezate et al. Reference Valdezate, Navarro, Villalón, Carrasco and Saéz-Nieto2010).
A cluster analysis of 16 E. ruminantium reference strains based on 12 VNTRs did not correlate with their geographical origins, except for subgroup C1, in which 4 strains from West Africa were grouped together. Although the PCA result also supported the splitting of cluster C from the remainder of the data set, there is the possibility that clustering of A and B might reflect a long-branch attraction artefact. Similarly, a poor association between genotypes and geographical origins of E. ruminantium strains has been demonstrated by MLST (Nakao et al. Reference Nakao, Magona, Zhou, Jongejan and Sugimoto2011). The migration of E. ruminantium from other endemic regions, presumably due to transportation of infected animals or ticks, may contribute to the genetic heterogeneity in a particular area. The rapid genome plasticity of E. ruminantium due to, for example, recombination between different genotypes (Collins et al. Reference Collins, Liebenberg, de Villiers, Brayton, Louw, Pretorius, Faber, van Heerden, Josemans, van Kleef, Steyn, van Strijp, Zweygarth, Jongejan, Maillard, Berthier, Botha, Joubert, Corton, Thomson, Allsopp and Allsopp2005; Frutos et al. Reference Frutos, Viari, Ferraz, Morgat, Eychenié, Kandassamy, Chantal, Bensaid, Coissac, Vachiery, Demaille and Martinez2006; Allsopp and Allsopp, Reference Allsopp and Allsopp2007; Hughes and French, Reference Hughes and French2007; Adakal et al. Reference Adakal, Meyer, Carasco-Lacombe, Pinarello, Allègre, Huber, Stachurski, Morand, Martinez, Lefrançois, Vachiery and Frutos2009; Nakao et al. Reference Nakao, Magona, Zhou, Jongejan and Sugimoto2011) may also play a significant role in the diversification of this bacterium and make it difficult to understand phylogenetic relationships between different genotypes.
The map1 PCR-RFLP and MLVA were used to genotype E. ruminantium harboured in field-collected A. variegatum. As previously reported by other researchers (Allsopp et al. Reference Allsopp, Dorfling, Maillard, Bensaid, Haydon, van Heerden and Allsopp2001; Martinez et al. Reference Martinez, Vachiéry, Stachurski, Kandassamy, Raliniaina, Aprelon and Gueye2004; Faburay et al. Reference Faburay, Geysen, Munstermann, Taoufik, Postigo and Jongejan2007b; Adakal et al. Reference Adakal, Stachurski, Konkobo, Zoungrana, Meyer, Pinarello, Aprelon, Marcelino, Alves, Martinez, Lefrancois and Vachiéry2010b), both methods demonstrated the circulation of different genotypes of E. ruminantium in heartwater endemic regions. However, since MLVA recovered 21 genotypes out of 38 ticks, MLVA showed greater discriminatory power compared to the map1 PCR-RFLP method, with which only 6 genotypes were obtained. Cluster analysis makes it possible to investigate the genetic relationships between each MLVA profile. MST analysis clearly showed that there are 2 major genotype clusters existing in Uganda, suggesting that the E. ruminantium population in this part of Uganda is dominated by a few clones that have undergone a degree of diversification resulting in discrete clonal clusters, as has been reported in Burkina Faso (Adakal et al. Reference Adakal, Gavotte, Stachurski, Konkobo, Henri, Zoungrana, Huber, Vachiery, Martinez, Morand and Frutos2010a). There was no association between MST groups and collection-sites of Ugandan field samples, which may indicate the occurrence of frequent movement of field isolates between these sampling sites. Five samples, MT-10, MT-13, MT-15, MT-16, and MT-19, were allocated to distinct positions. From our results it is difficult to ascertain whether the minor genotypes that were genetically distinct from 2 major clusters were recently introduced from other regions, or whether they had been maintained in the same areas but been segregated due to, for example, a lack of adaptation to the hosts or vectors. With reference to the development of region-specific vaccines, it becomes clear that genetic definition of the major local clonal clusters is an important requirement. However, there is also the possibility that the minor genotypes may result in outbreaks (or vaccine breakthrough), suggesting that it is necessary to monitor of the genetic makeup of populations over time.
The existence of non-pathogenic E. ruminantium variants (Allsopp et al. Reference Allsopp, van Strijp, Faber, Josemans and Allsopp2007) evokes the problematic issue of associating genotypes with pathogenicity in ruminants. The lack of knowledge on genetic determinants for virulence makes it difficult to distinguish pathogenic organisms from non-pathogenic ones using molecular genetic analysis such as PCR and sequencing. Thus, further studies on MLVA typing of isolates from heartwater cases and comparison with those circulating but not causing heartwater in the same region, which are maintained in the natural reservoir hosts and tick vectors, may help to disclose the pathogenic potential of each genotype.
A limited number of E. ruminantium strains were available to this study, and a lack of existing comprehensive information on the degree of cross-protection between strains means that it is difficult to draw conclusions with respect to correlation of MLVA genotypes with vaccine efficacy. However, the study raises some important points with respect to the use of MVLA. First, we have identified a panel of markers that will be highly useful tools for genotyping E. ruminantium isolates. Secondly, the Ugandan data suggest that in combination with clustering analysis, identification of genetic structure of E. ruminantium populations in the field is a distinct possibility. Additionally, the high discriminatory power as well as cost effective performance of MLVA provide the potential for this technique to be applied in the future with respect to optimizing vaccine trials by identifying local strain diversity, and also raise the possibility of exploring the association between E. ruminantium genotypes and phenotypes such as pathological outcome in the ruminant host.
ACKNOWLEDGEMENTS
We thank Mr T. Muroya and Ms A. Ohnuma for excellent technical assistance. We also appreciate two anonymous reviewers for their helpful comments.
FINANCIAL SUPPORT
The first author is supported by a research grant Fellowship from the Japanese Society for the Promotion of Science (JSPS) for young scientists. The second author is supported by a Royal Society University Research Fellowship. This work was supported by Grant-in-Aid for JSPS fellows and for Scientific Research from Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT), the program of Funding Research Center for Emerging and Re-emerging Infectious Disease, MEXT, and Asia-Africa S & T Strategic Cooperation Promotion Program by the Special Coordination Funds for Promoting Science & Technology, MEXT.















