Introduction
Camallanidae Railliet & Henry, 1915 is a specious family of nematodes infecting mainly fish (freshwater and marine), but also present in amphibians and reptiles (Anderson et al., Reference Anderson, Chabaud and Willmott2009). The systematics of this family has been widely debated; especially in relation to Procamallanus Baylis, 1923 and the validity of its subgenera (see Moravec and Sey, Reference Moravec and Sey1988; Moravec and Thatcher, Reference Moravec and Thatcher1997; Rigby and Adamson, Reference Rigby and Adamson1997; Anderson et al., Reference Anderson, Chabaud and Willmott2009; Moravec and Van As, Reference Moravec and Van As2015). Currently, the most accepted system for diagnosing genera (and/or subgenera) within Camallanidae is almost entirely based on the morphology of the buccal capsule (Moravec and Thatcher, Reference Moravec and Thatcher1997; Anderson et al., Reference Anderson, Chabaud and Willmott2009). However, genetic evidences suggest the artificiality of this system (Wijová et al., Reference Wijová, Moravec, Horák and Lukeš2006; Černotíková et al., Reference Černotíková, Horák and Moravec2011; Sardella et al., Reference Sardella, Pereira and Luque2017; Ailán-Choke et al., Reference Ailán-Choke, Davies, Tavares and Pereira2019, Reference Ailán-Choke, Tavares, Luque and Pereira2020).
Despite what is suggested by genetic analyses, such results are not sufficient to help resolve all the current taxonomic deadlocks observed in Camallanidae, as highlighted by Ailán-Choke et al. (Reference Ailán-Choke, Davies, Tavares and Pereira2019). Moreover, a scarce genetic database, sometimes containing inaccurate information, represents great challenge when dealing with the systematics of the family (Ailán-Choke et al., Reference Ailán-Choke, Tavares, Luque and Pereira2020). In order to overcome the taxonomic problems and the scarcity of genetic data, recent integrative taxonomic approaches have proven to be of great value for the systematic study of nematodes parasitic in vertebrates (e.g. Pereira and Luque, Reference Pereira and Luque2017, Pereira et al., Reference Pereira, Luque and Tavares2018; Ailán-Choke et al., Reference Ailán-Choke, Tavares, Luque and Pereira2020; Malta et al., Reference Malta, Paiva, Elisei, Tavares and Pereira2020).
Currently, there is no taxonomic study that formally integrates morphological, life history and genetic traits, as well as evaluates the importance of such data types, within the systematics of Camallanidae. Therefore, the current study had exactly this objective: data on three genetic markers [18S and 28S rDNA, cytochrome c oxidase subunit I (COI) mtDNA] and on important morphological and life history diagnostic traits (separated and concatenated), were used to reconstruct phylogenies including camallanids, in which the phylogenetic consistency and the mapping of traits were performed using bioinformatics. The main goals were: to evaluate the taxonomic consistency of genera/subgenera allocated in Camallanidae and their phylogenetic relationships, the characters that probably influence these relationships and their phylogenetic roles, and to compare the phylogenetic results of different datasets including different genetic markers, classical morphology and both combined. Additionally, new genetic sequences for Spirocamallanus pintoi Kohn & Fernandes, 1988 and for Spirocamallanus hilarii Vaz & Pereira, 1934 (a species currently uncharacterized genetically) are given.
Materials and methods
New genetic data
The following samples were used for genetic sequencing: one male of S. pintoi collected in Corydoras micracanthus Regan, 1912 (Siluriformes) from River La Caldera, and one male of P. S. hilarii collected in Astyanax endy Mirande, Aguilera & Azpelicueta, 2006 (Characiformes) from La Calderilla stream, localities belonging to the province of Salta, Argentina. The parasites were collected alive from freshly dead hosts, rinsed in saline and their mid-body excised and fixed in molecular grade 96–99% ethanol; the posterior and anterior body parts were fixed in hot 4% formaldehyde solution and stored in 70% ethanol for morphological identification. Other specimens from the same infrapopulations were also fixed and processed for confirming the morphological identification. Voucher specimens were deposited in the Parasitological Collection of the Museo Argentino de Ciencias Naturales ‘Bernardino Rivadavia’, Buenos Aires, Argentina (MACN-Pa) (accession nos. S. pintoi: MACN-Pa 751; S. hilarii: MACN-Pa 752). All procedures involving animal manipulation were approved by the Secretaría de Medio Ambiente, Ministerio de Ambiente y Producción Sustentable, Gobierno de la provincia de Salta, Argentina (Authorization No. 000248/14), and were strictly according to the international ethical standards in animal research. Protocols related to polymerase chain reaction, use of primers and sequencing were those described by Ailán-Choke et al. (Reference Ailán-Choke, Tavares, Luque and Pereira2020), targeting partial fragments of the 18S and 28S rDNA, and COI mtDNA. Contiguous sequences were assembled in Geneious (Geneious ver. 9, created by Biomatters), their consensus extracted and deposited in GenBank (see ‘Results’ section). Preliminary BLAST search (https://www.ncbi.nlm.nih.gov/genbank/) was performed in order to confirm the genetic proximity between the present samples and other camallanids.
Phylogenetic analysis of genetic data
For phylogenetic reconstructions based on genetic data, we considered three separated datasets according to the targeted genetic regions (18S, 28S and COI). Alignments of different genes were not concatenated due to the lack of correspondence between sampled taxa, and in order to improve sampling coverage (taxa diversity). In addition to the newly obtained sequences, others were retrieved from GenBank database according to the following criteria: taxa identified at the species level and allocated in Camallanidae, with full information on host species and geographic origin, same genetic coverage as in the present sequences and with minimum length of 818 bp for 18S, 463 bp for 28S and 339 bp for COI, and from which taxonomic descriptions had most of the characters considered here well detailed (see as follows). Because of the numerous similar sequences for the same gene from the same species (clones and isolates), and the apparent misidentifications in the genetic database related to camallanids (see Wijová et al., Reference Wijová, Moravec, Horák and Lukeš2006; Černotíková et al., Reference Černotíková, Horák and Moravec2011; Ailán-Choke et al., Reference Ailán-Choke, Davies, Tavares and Pereira2019, Reference Ailán-Choke, Tavares, Luque and Pereira2020), only one representative of each specific taxa was included, in which this representative was chosen based on previous studies indicating the correct taxonomic identification of the sample (Ailán-Choke et al., Reference Ailán-Choke, Davies, Tavares and Pereira2019, Reference Ailán-Choke, Tavares, Luque and Pereira2020). These precautions were taken to prevent inaccurate results and all the sequences used in the current study, along with information on host, geographic origin, GenBank accession numbers and references used for species morphological description, are listed in Table 1. The outgroup was chosen according to previous phylogenies of Camallanidae (Sardella et al., Reference Sardella, Pereira and Luque2017; Ailán-Choke et al., Reference Ailán-Choke, Davies, Tavares and Pereira2019, Reference Ailán-Choke, Tavares, Luque and Pereira2020).
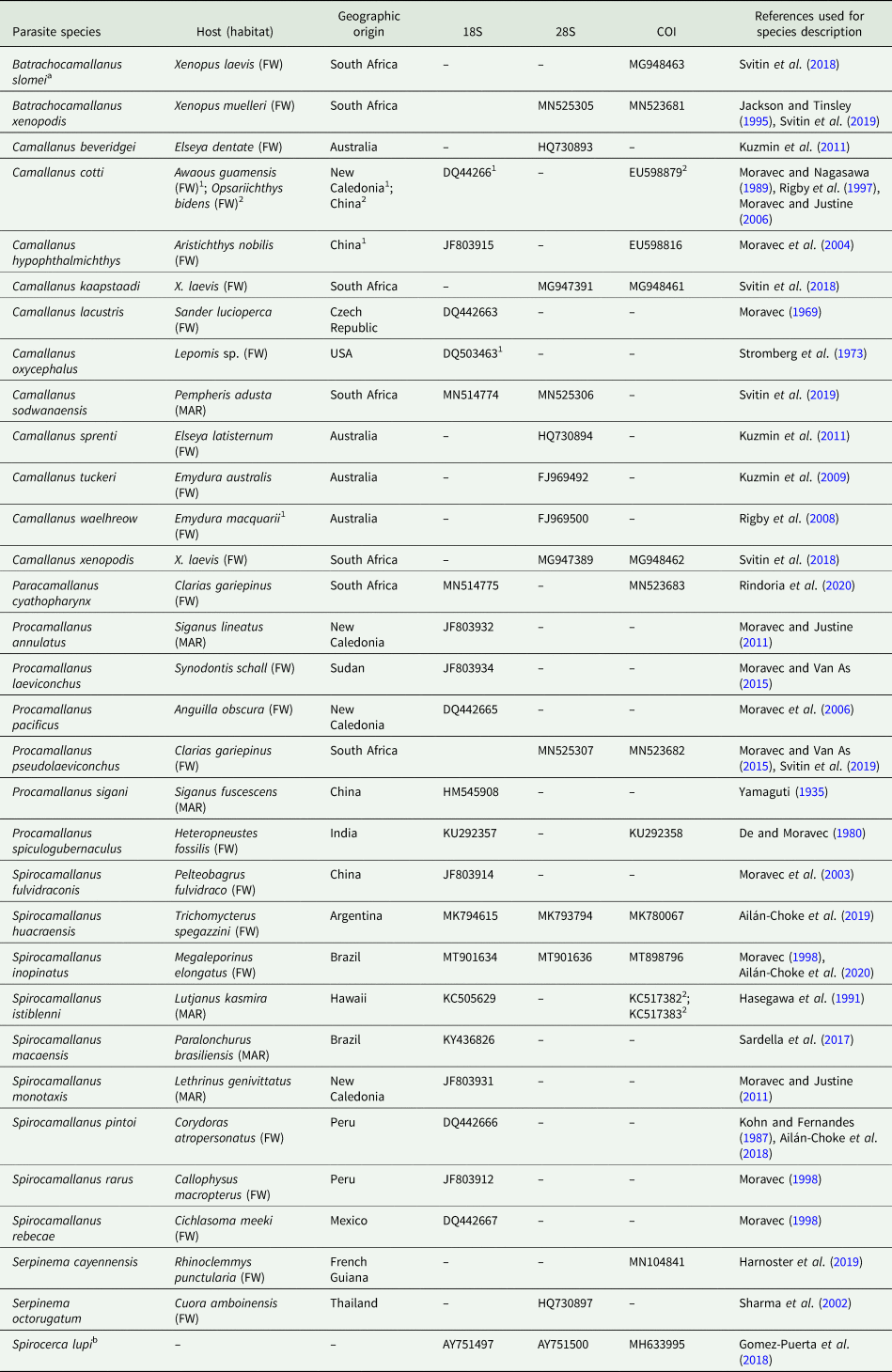
Table 1. Parasite species used in the current study, associated with host [habitat: freshwater (FW) or marine (MAR)] and geographic origin corresponding to the isolates that were used for genetic sequencing
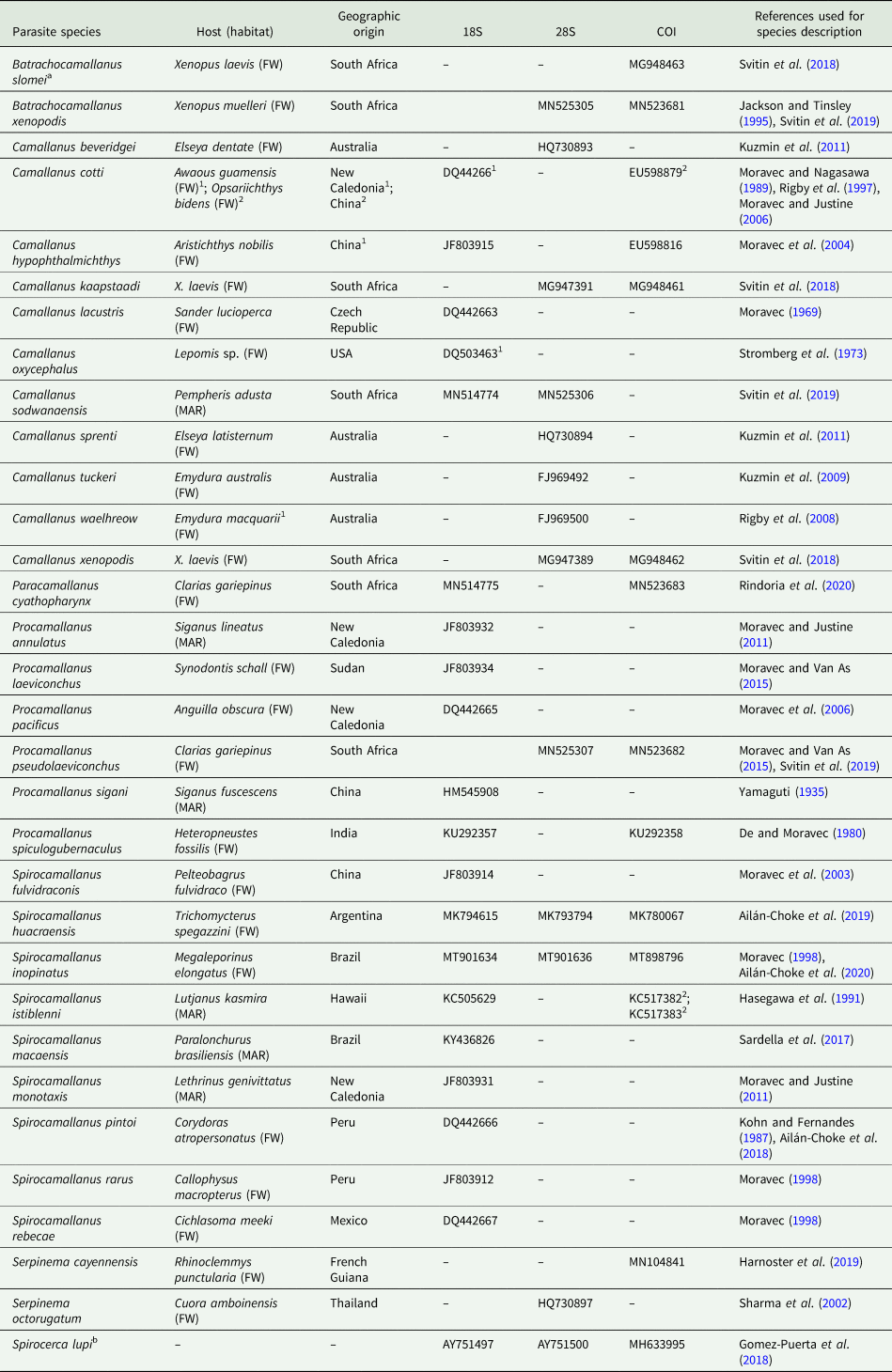
GenBank accession numbers, according to each genetic marker, and reference(s) used for morphological characterization are also given.
a Referred to as Procamallanus slomei in GenBank.
b Used as the outgroup in the phylogenetic analyses.
The 18S and 28S refer to the SSU and LSU of the rRNA gene, respectively, and the COI to the cytochrome c oxidase subunit I mtDNA. Superscript numbers make correspondence between information.
Sequences were aligned using the multiple algorithm tool M-Coffee (Notredame et al., Reference Notredame, Higgins and Heringa2000), then submitted to the transitive consistency score, to verify the reliability of aligned positions and, those scored as averaged to bad were automatically trimmed (Chang et al., Reference Chang, Di Tommaso and Notredame2014). Saturation of nucleotide substitution in the alignments was also evaluated using Xia's method, implemented in software Data Analysis in Molecular Biology and Evolution (DAMBE), to confirm their adequateness for phylogenetic reconstructions (Xia et al., Reference Xia, Xie and Li2003; Xia, Reference Xia2018). Alignments were subjected to the Automatic Barcode Gap Discovery online (http://wwwabi.snv.jussieu.fr/public/abgd/), to estimate the genetic distances among the sampled sequences (18S, 28S and COI), using Kimura two-parameter (K2P) as the distance metric (Kimura, Reference Kimura1980; Puillandre et al., Reference Puillandre, Lambert, Brouillet and Achaz2012).
Based on the advantages shown by Bayesian inference in recent integrative taxonomic studies (e.g. Hernández-Lara et al., Reference Hernández-Lara, Espinosa de los Monteros, Ibarra-Cerdeña, García-Feria and Santiago-Alarcon2018; Sayad and Yassin, Reference Sayad and Yassin2019; van der Wal et al., Reference van der Wal, Ahyong, Ho, Lins and Lo2019), all phylogenetic hypothesis were estimated using this approach in BEAST 2.5 (Bouckaert et al., Reference Bouckaert, Vaughan, Barido-Sottani, Duchêne, Fourment, Gavryushkina, Heled, Jones, Kühnert, De Maio, Matschiner, Mendes, Müller, Ogilvie, Du Plessis, Popinga, Rambaut, Rasmussen, Siveroni, Suchard, Wu, Xie, Zhang, Stadler and Drummond2019); the best-fit substitution model for each dataset was chosen according to bModelTest (Bouckaert and Drummond, Reference Bouckaert and Drummond2017), the molecular clock model was relaxed (log normal), defined using the nested sampling method (Russel et al., Reference Russel, Brewer, Klaere and Bouckaert2019) and the Yule tree prior, selected according to the posterior densities and the effective sample sizes (ESS), calculated in Tracer (Rambaudt et al., Reference Rambaudt, Drummond, Xie, Baele and Suchard2018). This approach was preferred by its robustness, because it provides improved evolutionary pathways in phylogenetic reconstruction without overestimating nodal supports (Bouckaert et al., Reference Bouckaert, Vaughan, Barido-Sottani, Duchêne, Fourment, Gavryushkina, Heled, Jones, Kühnert, De Maio, Matschiner, Mendes, Müller, Ogilvie, Du Plessis, Popinga, Rambaut, Rasmussen, Siveroni, Suchard, Wu, Xie, Zhang, Stadler and Drummond2019), and in order to prevent biased comparative results between different data types. The posterior estimates of parameter densities and the ESS for each parameter, as well as the posterior probability for nodal supports in the majority rule consensus phylogenetic trees, were determined after running the Markov chain Monte Carlo (MCMC), always four chains in two runs, each run with number of generations varying from 10 × 106 to 20 × 106 in order to reach chain convergence, 25% burn-in and saving the last 10 001 trees. The quality of the analysis (parameter densities, ESS and burn-in) and the chain convergence were examined in Tracer (Rambaudt et al., Reference Rambaudt, Drummond, Xie, Baele and Suchard2018).
The genetic markers’ profile for phylogenetic informativeness was also evaluated using PhyDesign (Townsend, Reference Townsend2007), based on the phylogenetic trees generated from genetic data.
Morphological data, systematic classification and morphology-based phylogeny
The proposal of Moravec and Thatcher (Reference Moravec and Thatcher1997) validating Procamallanus and Spirocamallanus as subgenera of Procamallanus, that of Petter (Reference Petter1979) validating Procamallanus and Spirocamallanus as genera, that of Jackson and Tinsley (Reference Jackson and Tinsley1995) validating Batrachocamallanus Jackson & Tinsley, Reference Jackson and Tinsley1995, and that of Anderson et al. (Reference Anderson, Chabaud and Willmott2009) validating the remaining genera were evaluated herein. Based on these previously mentioned studies and the numerous species descriptions used in the current study (see Table 1), we used software Mesquite (Maddison and Maddison, Reference Maddison and Maddison2019) to generate a matrix with 20 characters and 88 states in total, which are detailed in Supplementary material 1. A summary of the characters and their number of states are summarized as follows: (1) number of cephalic papillae (three states), (2) presence of sclerotized tridents associated with buccal capsule (two states), (3) buccal capsule structure (three states), (4) buccal capsule ridges (four states), (5) laminar teeth at base of buccal capsule (two states), (6) relative position of deirids (three states), (7) relative position of excretory pore (three states), (8) relative position of vulva in females (three states), (9) protrusion of vulval lips (two states), (10) general morphology of tail in females (two states), (11) number of caudal spikes on tail in females (four states), (12) presence of caudal alae in males (two states), (13) number of caudal spikes on tail in males (four states), (14) presence of pedunculate papillae in males (two states), (15) presence of adcloacal papillae in males (two states), (16) spicule symmetry in males (two states), (17) presence of gubernaculum in males (two states), (18) habitat (two states), (19) host order (20 states) and (20) biogeographic occurrence (11 states). Characters and states were all discrete, with the same weight (there is no previous study permitting the inference of character weighing); states were all coded with numbers (0–9), except for in the cases of ‘host order’ and ‘biogeographic occurrence’, which were the only with polymorphic states and thus, additional letters (A–J) had to be used for coding; missing data were coded with a question mark (?). In all characters, an additional ‘null’ state, different from all others, was intentionally added for the outgroup, in order to polarize the ingroup Camallanidae, since the monophyly of the family is clear and confirmed (Wijová et al., Reference Wijová, Moravec, Horák and Lukeš2006; Černotíková et al., Reference Černotíková, Horák and Moravec2011; Ailán-Choke et al., Reference Ailán-Choke, Davies, Tavares and Pereira2019, Reference Ailán-Choke, Tavares, Luque and Pereira2020).
Nomenclature and classification of hosts were updated following Froese and Pauly (Reference Froese and Pauly2021), Frost (Reference Frost2021) and Uetz et al. (Reference Uetz, Freed and Hosek2020). Freshwater, marine and terrestrial biogeographic regions follow Olson et al. (Reference Olson, Dinerstein, Wikramanayake, Burgess, Powell, Underwood, D'Amico, Itoua, Strand, Morrison, Loucks, Allnutt, Ricketts, Kura, Lamoreux, Wettengel, Hedao and Kenneth2001) and Kocsis et al. (Reference Kocsis, Reddin and Kiessling2018).
Taxa of the matrix containing morphological and life history traits (from here, generalized as morphological data) were edited to match those from each genetic marker (18S, 28S and COI). Then, the matrices were subjected to Bayesian inference as previously described, but under the Markovian Mkv model of character change (Lewis, Reference Lewis2001), using Morph-models package implemented in BEAST 2.5 (Bouckaert et al., Reference Bouckaert, Vaughan, Barido-Sottani, Duchêne, Fourment, Gavryushkina, Heled, Jones, Kühnert, De Maio, Matschiner, Mendes, Müller, Ogilvie, Du Plessis, Popinga, Rambaut, Rasmussen, Siveroni, Suchard, Wu, Xie, Zhang, Stadler and Drummond2019).
Integrated analysis of morphological, life history traits and genetic data, character evaluation and mapping
The morphological data matrices were manually concatenated with the three genetic alignments, resulting in three datasets with two partitions each (from here, referred to as concatenated data): morphology + 18S, morphology + 28S and morphology + COI. The phylogenies were reconstructed using Bayesian inference in BEAST 2.5 as previously described, with clock model, tree priors and MCMC parameters linked between the partitions and substitution model unlinked. In software Phylogenetic Analysis Using Parsimony (PAUP), the consistency indexes (CIs) of morphological characters were estimated to measure their degree of homoplasy (phylogenetic consistency), in which 0 < CI < 1, where CI = 0 indicates full homoplasy and CI = 1 indicates lack of homoplasy; CI ⩽ 0.6 generally indicates significant presence of homoplasy and low consistency (Swofford, Reference Swofford2002). Characters and states were mapped in the majority rule consensus tree using software Mesquite (Maddison and Maddison, Reference Maddison and Maddison2019). Consistency of phylogenetic trees (from genetic, morphological and concatenated data) was also evaluated by their CIs as previously described.
Statistical analysis for evaluating differences in the consistency of phylogenies and in the morphological characters
For comparing the consistency of different phylogenetic approaches, we calculated the CIs of the last 10 001 trees generated from genetic, morphological and concatenated data, according the three datasets previously mentioned (18S, 28S and COI). Within each dataset, differences in the CIs of these trees were tested, as well as the CI of the 20 morphological characters between morphological vs concatenated trees, using the non-parametric Friedman test for paired data (Zar, Reference Zar2010) (data were paired with to respect the order of generations in the MCMC). CI differences among 18S vs 28S vs COI datasets were not tested, because results could be biased due to different taxa configuration. Post-hoc Wilcoxon test for paired data, with Holm–Bonferroni P-adjustment method, was used when the explanatory variable had more than two categories (Zar, Reference Zar2010). Non-parametric inferential statistic was performed because CI values, in all occasions, were not normally distributed and not homoscedastic (Zar, Reference Zar2010).
Results
Newly obtained sequences and their comparison with other camallanids
Partial sequences of the 18S (867 bp/MW930855), 28S (875 bp/MW930866) and COI (388 bp/MW930301) for S. hilarii and those of the 28S (837 bp/MW930867) and COI (388 bp/MW930302) for S. pintoi were obtained for the first time; in addition to a newly obtained partial sequence of the 18S (791 bp/MW930855) for S. pintoi from different host and locality. The genetic distances (indicated as K2P) between the present sequences were 0.014, 0.130 and 0.224 for the 18S, 28S and COI, respectively. BLAST search showed that 18S sequences of both S. hilarii and S. pintoi were most similar to that of S. pintoi (DQ442666), isolated in Corydoras atropersonatus Weitzman & Nijssen, 1970 (Siluriformes) from Peru (raw genetic similarities 98.9 and 99.7%, respectively). Genetic similarity of 28S sequences of S. hilarii and S. pintoi were higher in relation to S. inopinatus Travassos, Artiga & Pereira, 1928 (MT901638), isolated from Anostomoides passionis Santos & Zuanon, 2006 (Characiformes), in Brazil (raw genetic similarities 65.6 and 66.5%, respectively). The COI sequence of S. hilarii was most similar to the same isolate of S. inopinatus (MT898797) (raw genetic similarity 83.4%) previously cited, and the COI sequence of S. pintoi most similar to that of Camallanus kaapstaadi Southwell & Kirshner, 1937 (MG948461) isolated in Xenopus laevis (Daudin, 1802) (Anura), from South Africa (raw genetic similarity 84.7%). The patterns of K2P distances from nuclear markers were similar to each other, and slightly different from that of the mitochondrial marker; in the dataset of 18S and 28S, the present sequences of S. hilarii and S. pintoi were closer together (18S K2P = 0.014; 28S K2P = 0.130), and S. inopinatus (MT901635/MT901638) the next closest (18S K2P = 0.015/0.007; 28S K2P = 0.134/0.125). In contrast, K2P distances in the COI dataset indicated close relatedness of S. istiblenni (Noble, 1966) (KC517383), isolated in Lutjanus kasmira (Forsskål, 1775) (Perciformes) from Hawaii, with S. hilarii (K2P = 0.149) and S. pintoi (K2P = 0.187), as well as between S. hilarii and S. inopinatus (K2P = 0.187), and between S. pintoi and C. kaapstaadi (K2P = 0.167) (for the complete matrices see Supplementary material 2).
Comparison between phylogenies inferred from different datasets and approaches
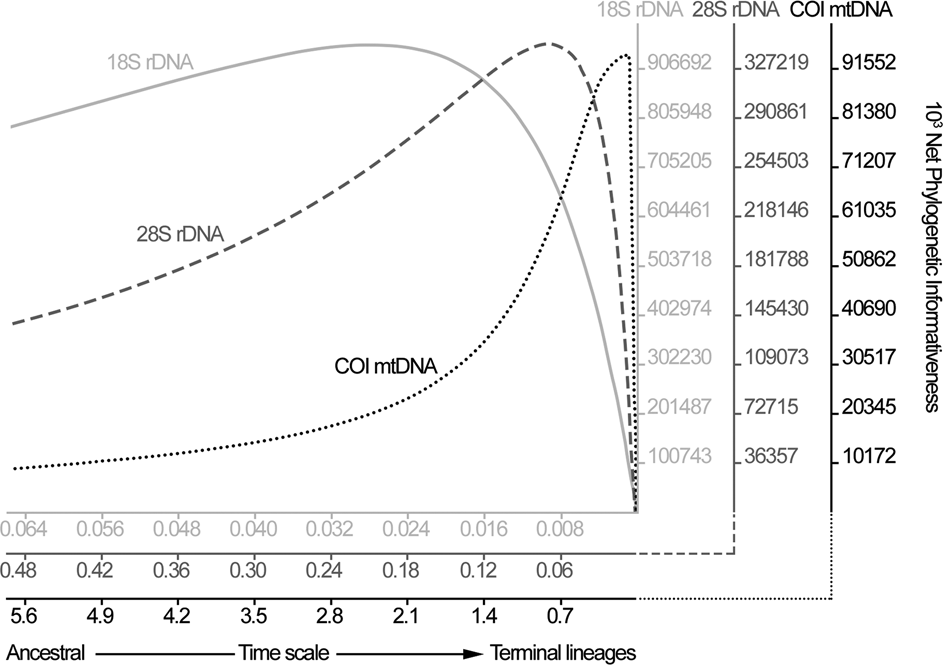
The phylogenetic informativeness of genetic markers indicated a gradual scale, in which 18S was better informative for divergence events related to deeper nodes, reducing its power as the time scale approached to the terminal lineages. Reciprocally, COI was more informative closer to the terminal lineages and the 28S exhibited an intermediate pattern between that of 18S and COI (Fig. 1). Xia's test indicated no substantial saturation in the alignments; observed values of the index of substitution saturation were all statistically (P < 0.001) lower than the critical values (Iss < Iss.c).
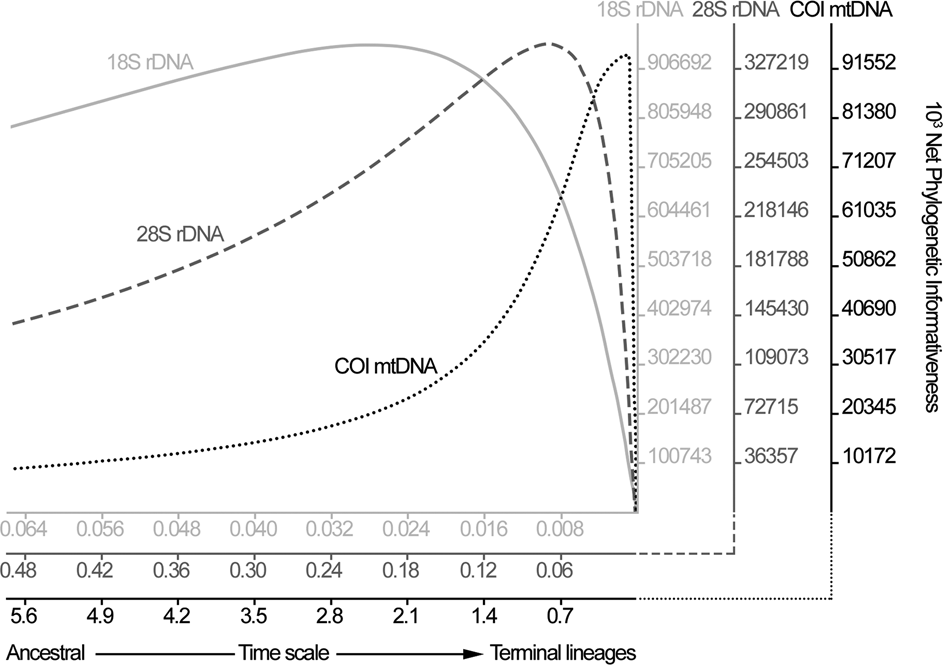
Fig. 1. Graphical representation of phylogenetic informativeness for each genetic marker used in the current study, estimated by PhyDesign.
Taxa coverage was considerably different between the datasets. None of the Camallanus Railliet & Henry, 1915 species in the 18S analysis were to be found in the 28S analysis. There were five species of Procamallanus and 10 species of Spirocamallanus Olsen, 1952 in the 18S analysis and only one of Procamallanus and four of Spirocamallanus in the 28S analysis. The COI analysis was the only including a species of Paracamallanus; one part of the remaining taxa was present in the 18S analysis, and the other included in that of the 28S.
The topology of the phylogenies inferred from morphological, genetic and concatenated data showed more similarities than divergences; the phylogenies of COI dataset were more divergent than those of 18S and 28S (Figs 2–4; and see Supplementary material 3 for morphological trees). In this sense, only in COI dataset, the CI of the tree inferred from morphological data (CI = 0.702) was statistically (P = 0.01) higher than those from the trees inferred from genetic and concatenated data (CI = 0.542 and 0.549, respectively); CIs of genetic and concatenated data trees from COI dataset were the lowest (Table 2). In general, the concatenated data seemed to return phylogenetic information and tree topology slightly improved (Figs 2–4).

Fig. 2. Phylogenetic trees of genetic (upper tree) and concatenated (bottom tree) data, generated using Bayesian inference. Nodal supports are based on Bayesian posterior probability (BPP) and represented as follows: full squares (BPP = 1), empty squares (0.96 < BPP < 1), full circles (0.94 < BPP <0 .96) and empty circles (0.90 < BPP < 0.94). Main morphological and life history traits are depicted and mapped in the concatenated data tree labelled as follows: A (articulated buccal capsule with sclerotized tridents associated and longitudinal ridges not separated into dorsal and ventral groups), B (buccal capsule, not articulated, with spiral ridges and laminar teeth at its base), C (deirids between buccal capsule base and nerve ring), D and E (male with caudal alae, pedunculate papillae, asymmetric spicules, gubernaculum absent and present, respectively) and F (male without caudal alae, with sessile papillae and symmetric spicules). Question marks indicate unknown state for the position of deirids. Habitat, host taxa and geographic origin are those of the isolate genetic sequenced; the superscript asterisks indicate polymorphic states for the character. Male of Procamallanus pacificus is unknown. Sequences from the current study are in bold.
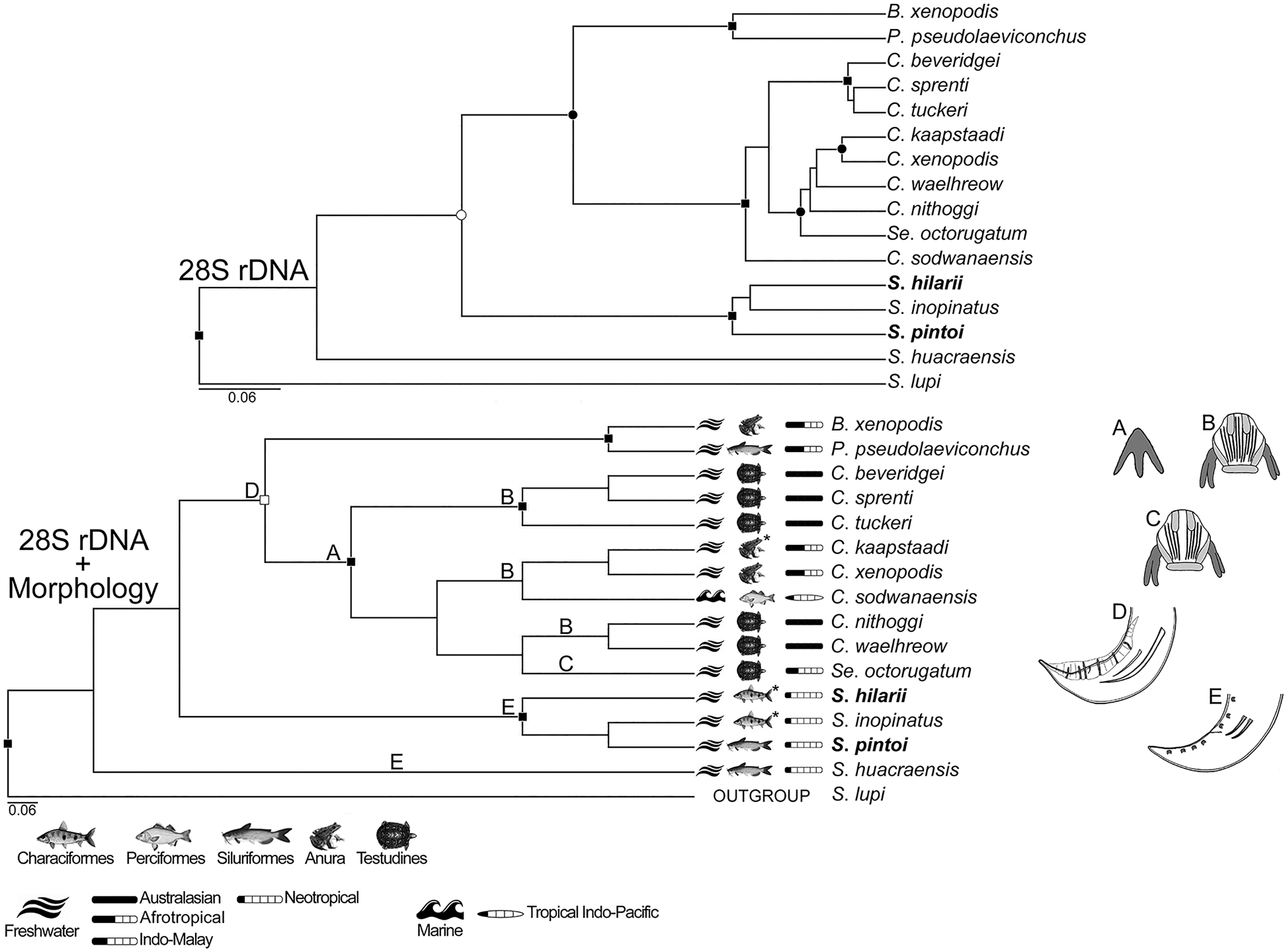
Fig. 3. Phylogenetic trees of genetic (upper tree) and concatenated (bottom tree) data, generated using Bayesian inference. Nodal supports are based on Bayesian posterior probability (BPP) and represented as follows: full squares (BPP = 1), empty squares (0.96 < BPP < 1), full circles (0.94 < BPP < 0.96) and empty circles (0.90 < BPP < 0.94). Main morphological and life history traits are depicted and mapped in the concatenated data tree labelled as follows: A (sclerotized tridents associated with buccal capsule present), B and C (articulated buccal capsule with longitudinal ridges not and separated into dorsal and ventral groups, respectively), D (male with caudal alae, pedunculate papillae, asymmetric spicules, gubernaculum absent) and E (male without caudal alae, with sessile papillae and symmetric spicules). Habitat, host taxa and geographic origin are those of the isolate genetic sequenced; the superscript asterisks indicate polymorphic states for the character. Sequences from the current study are in bold.
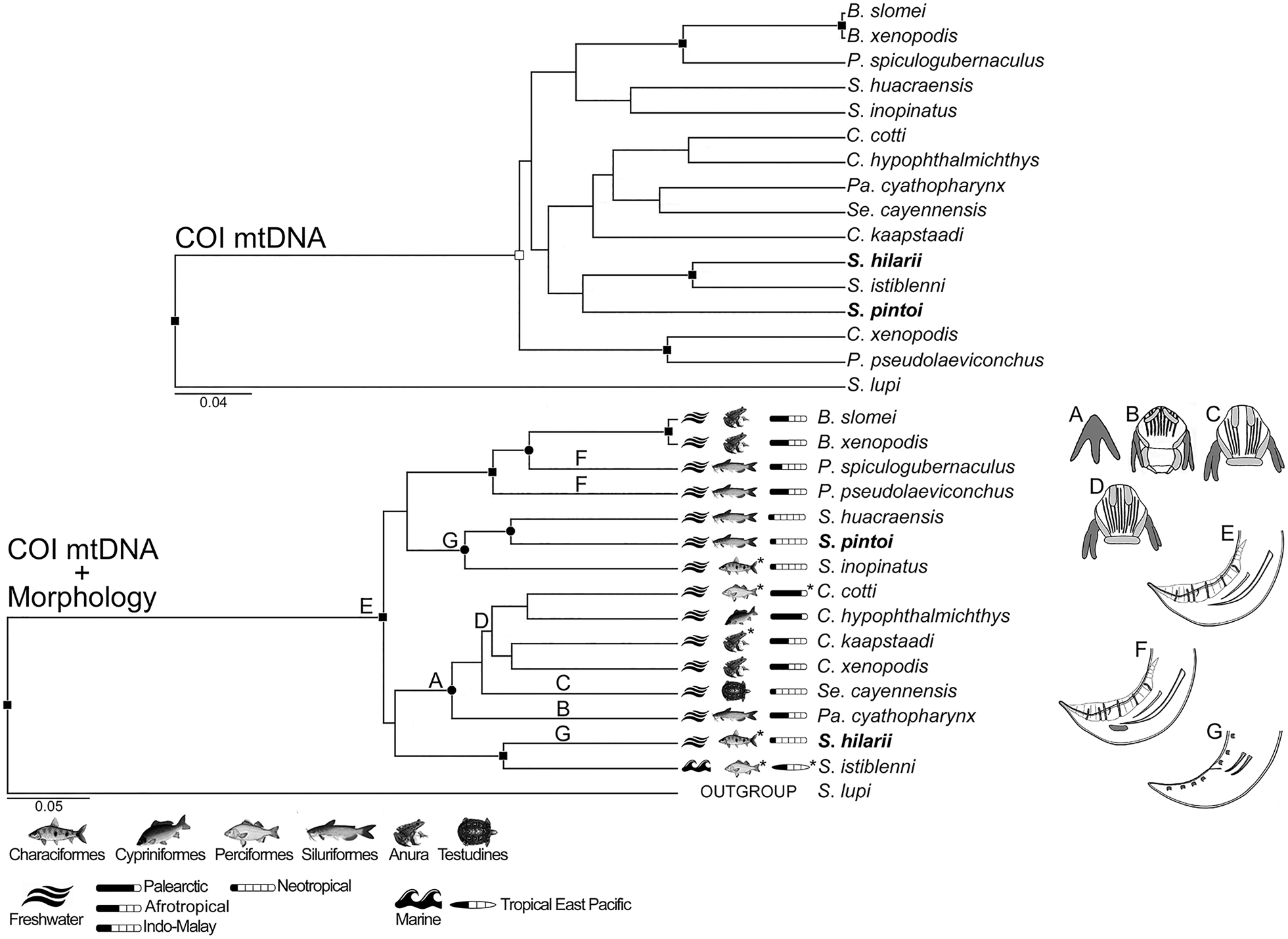
Fig. 4. Phylogenetic trees of genetic (upper tree) and concatenated (bottom tree) data, generated using Bayesian inference. Nodal supports are based on Bayesian posterior probability (BPP) and represented as follows: full squares (BPP = 1), empty squares (0.96 < BPP < 1) and full circles (0.94 < BPP < 0.96). Main morphological and life history traits are depicted and mapped in the concatenated data tree labelled as follows: A (sclerotized tridents associated with buccal capsule present), B (articulated buccal capsule, with longitudinal ridges and distinct basal cavity), C and D (articulated buccal capsule with longitudinal ridges not and separated into dorsal and ventral groups, respectively), E and F (male with caudal alae, pedunculate papillae, asymmetric spicules, gubernaculum absent and present, respectively) and G (male without caudal alae, with sessile papillae and symmetric spicules). Habitat, host taxa and geographic origin are those of the isolate genetic sequenced; the superscript asterisks indicate polymorphic states for the character. Sequences from the current study are in bold.
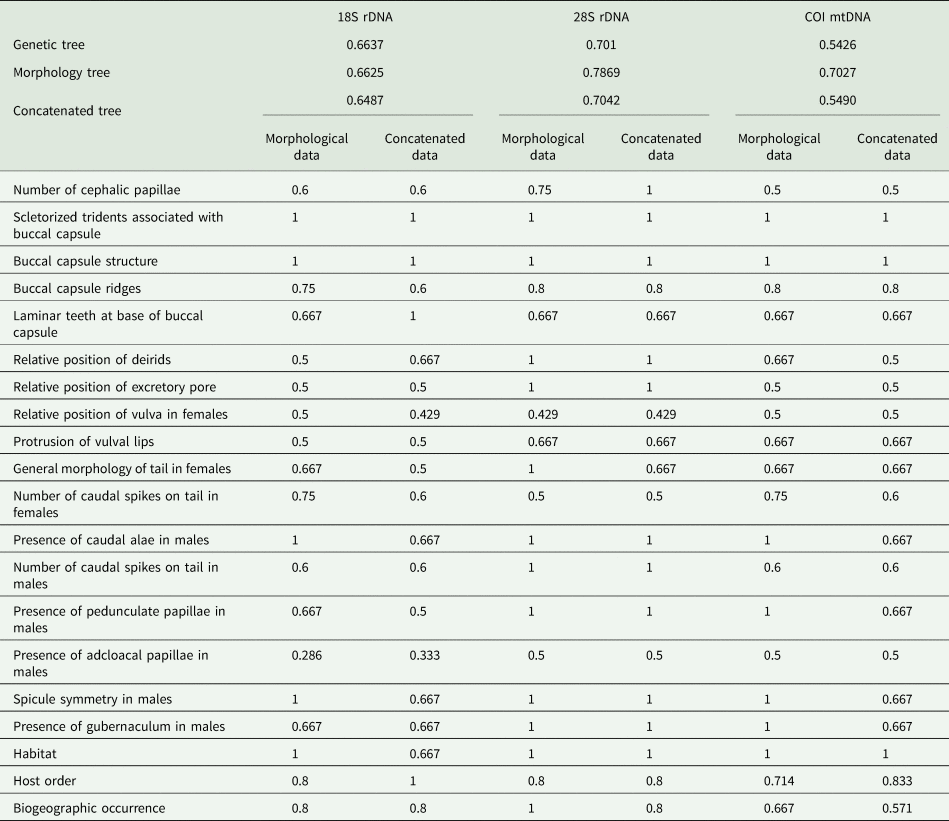
Table 2. CI values for phylogenetic trees, morphological and life history traits, related to camallanids from the current study, according to each dataset
Spirocamallanus included the highest number of representatives and was not monophyletic in all phylogenies, forming full to high-supported assemblages, which diverged independently in different times, being better observed in the 18S dataset (Fig. 2). Similar patterns were observed in Procamallanus. Exceptions were only found in the trees generated from morphological data in 28S and COI datasets, in which these subgenera appeared as monophyletic (Supplementary material 3). The second most diverse taxon was Camallanus that appeared as a fully supported monophyletic assemblage in the trees from 18S dataset, and in the concatenated tree of COI dataset, but with low support in the latter (Figs 2 and 4). In the genetic and concatenated trees from 28S dataset, representatives of Camallanus formed a fully supported assemblage; however, with the presence of Serpinema Yeh, 1960 within this clade, Camallanus was not monophyletic (Fig. 3). Batrachocamallanus was represented by one and two taxa in the datasets of 28S and COI, respectively, forming a full-supported monophyletic group, which was sister lineage of Procamallanus spp. with full support (Figs 3 and 4). Camallanus, Paracamallanus and Serpinema appeared closely related, sharing a common recent ancestor with full to high nodal supports in the phylogenies, except in the genetic data tree of COI, where supports were low, and C. xenopodis Jackson & Tinsley, Reference Jackson and Tinsley1995 (MG948462) formed a basal fully supported group with Procamallanus pseudolaeviconchus Moravec & Van As, Reference Moravec and Van As2015 (MN523682) (Figs 3 and 4).
Evaluation of morphological and life history traits, and character mapping in concatenated data trees
CI values of morphological characters (also referring to life history traits) were statistically similar between the phylogenies inferred from morphological and concatenated data, in all datasets; even though values tended to be higher using morphological data only, because the tendency in this type of dataset is the overestimation of morphological characters (Table 2). Some of the most consistent characters were the ‘sclerotized trident associated with buccal capsule’ related to the presence/absence of two cuticular tridents associated with dorsal and ventral sides of buccal capsule, and the ‘buccal capsule structure’ that could not be articulated, composed of two articulated valves with no basal cavity, or composed of two articulated valves with basal cavity (Table 2; Supplementary material 1). These characters, along with ‘buccal capsule ridges’ that is related to the presence/absence and orientation of cuticular ridges on the buccal capsule wall, were congruent with the lineages of Camallanus, Paracamallanus and Serpinema; as a common and exclusive trait, these taxa shared the presence of the buccal capsule tridents (Figs 2–4). All these three previously mentioned characters, showed lack or low degree of homoplasy, except for in the case of the ‘buccal capsule ridges’, in which CI tended to be low (i.e. 0.6) in the 18S dataset (Table 2). It is important to highlight that buccal capsule ridges were not consistent with the phylogenetic assemblages of species of Procamallanus and Spirocamallanus, the most numerous taxa in the 18S dataset (Fig. 2). Nevertheless, regarding the buccal capsule morphology, S. huacraensis Ramallo, 2008 and Spirocamallanus rarus Travassos, Artigas & Pereira, 1928 formed a fully supported clade, sharing the presence of ‘laminar teeth at base of buccal capsule’ (Fig. 2), being observed only in the 18S dataset showing lack or low degree of homoplasy (Table 2); this trait was present only in these two species and there are no sequences other than 18S available for S. rarus.
The morphological features present in the posterior end of males were also consistent with some highly to fully supported assemblages; these features included the presence/absence of caudal alae, pedunculate papillae and gubernaculum, as well as the asymmetrical spicule (Figs 2–4). CIs of these characters were all higher than 0.6 (Table 2). Most of the taxa analysed have males with broad caudal alae, pedunculate papillae and spicules asymmetric; the presence of gubernaculum in these representatives was shared by the group consisting of Procamallanus laeviconchus Wedl, 1862, Procamallanus pacificus Moravec, Justine, Würtz, Taraschewski & Sasal, Reference Moravec, Justine, Würtz, Taraschewski and Sasal2006, Procamallanus spiculogubernaculus Agarwal, 1958 and Spirocamallanus fulvidraconis Li, 1935, which was moderately supported in the genetic data tree of 18S (Fig. 2), and also observed in the concatenated data tree of COI, in two closely related lineages, i.e. that of P. spiculogubernaculus and P. pseudolaeviconchus (Fig. 4). In contrast, S. hilarii, S. inopinatus and S. pintoi, which have males lacking caudal alae, pedunculate papillae and with symmetric spicules, formed fully supported groups in the trees of 18S and 28S datasets (Figs 2 and 3). However, in the trees of COI dataset, S. huacraensis replaced S. hilarii forming a moderate-supported assemblage with S. inopinatus and S. pintoi (Fig. 4). Spirocamallanus hilarii, S. inopinatus and S. pintoi shared similar caudal morphology in males and formed fully supported assemblages, except when COI sequences were included (Figs 2–4). Spirocamallanus huacraensis that has males with similar caudal structure to S. hilarii, S. inopinatus and S. pintoi, was some phylogenetic related to these species; however, nodal supports illustrating these relationships were low and the phylogenetic position of S. huacraensis variable (Figs 2–4).
The position of deirids, between the base of buccal capsule and anterior to nerve ring, was shared by a clade formed by several species of Procamallanus and Spirocamallanus, with moderate and high supports, in the genetic and concatenated data trees of 18S dataset, respectively (Fig. 2). This feature was also present in the highly supported clade of S. inopinatus and S. pintoi in the same dataset (Fig. 2). The CI of this character was improved in the concatenated data tree, when compared to that of morphological tree (from 0.5 to 0.667), although with no statistical difference.
Habitat and host order were also consistent with the phylogenetic topologies; the related CIs were all higher than 0.667 (Table 2). However, habitat should be interpreted with caution because most taxa were from freshwater, and 18S dataset was the only one including more than one taxa from marine environments (Figs 2–4). Nevertheless, species from marine habitats were assembled together forming moderate and highly supported clades in genetic and concatenated data trees, respectively, except for by Spirocamallanus rebecae Andrade-Salas, Pineda-López & García-Magaña, 1994 (Fig. 2). Similarities on host order was also a pattern observed within and among closely related lineages; this character was consistent (CIs ⩾0.8, see also Table 2) with the phylogenetic hypotheses of concatenated data, and the pattern was similar in the genetic data trees of all datasets (Figs 2–4). Most representatives of Camallanidae were isolated from fish belonging to Siluriformes and Perciformes, in which these orders along with Characiformes, were similar in closely related lineages (highly to fully-supported), being better observed in the 18S dataset trees (Figs 2–4). Amphibian hosts allocated in Anura were common in full supported assemblage of Batrachocamallanus, but also present in some lineages of Camallanus (Figs 3 and 4). Patterns on reptile hosts of the order Testudines could be observed only on the 28S dataset, because it included more than one representative with this character and state, in which it was shared by some assemblages of Camallanus, in addition to Serpinema (Fig. 3). It should be mentioned that in the genetic tree of the 28S dataset, Camallanus sodwanaensis Svitin, Truter, Kudlai, Smit & du Perez, Reference Svitin, Truter, Kudlai, Smit and du Preez2019, the only Camallanus isolated from fish, formed an external basal lineage to a clade formed by Serpinema and other Camallanus species, all parasitic in amphibian and reptile hosts (Fig. 3).
The patterns observed for the character ‘biogeographic occurrence’ were similar to those of host order. CIs for this character was generally high, except for in the tree of concatenated data from COI in that it tended to be low (Table 2). Biogeographic similarities were clearer in the assemblages of species from freshwater environment (Figs 2–4). These patterns were also better illustrated by the 18S dataset phylogenies, in which species from the Neotropical region formed fully supported and somewhat closely related clades; marine species from Indo and East Pacific were phylogenetically close as well (Fig. 2). The analysis including 28S sequences also indicated close relatedness among species from the Neotropical region; however, S. huacraensis was out of the ‘Neotropical assemblage’ (Fig. 3). Similarly, the concatenated data tree using COI sequences indicated phylogenetic proximity between Spirocamallanus species from the Neotropical region, with the exception of S. hilarii (Fig. 4). It was also possible to observe in the 28S dataset that, species of Camallanus from Australasian region formed full and low supported inner assemblages, in which these lineages kept some phylogenetic relationship (Fig. 3). Outside the clade including species of Camallanus, Paracamallanus and Serpinema, in the phylogenetic hypotheses from 28S and COI datasets, samples from the Afrotropical region represented by Batrachocamallanus spp. and P. pseudolaeviconchus were grouped together with full support, and they were close to P. spiculogubernaculus from Indo–Malayan region (Figs 3 and 4).
Some species used in the current study showed a wide host range, being reported in hosts from different orders in some cases [e.g., Camallanus cotti Fujita, 1927, Camallanus lacustris (Zoega, 1776) and Camallanus oxycephalus Ward & Magath, 1916]; for these species ‘host order’ was highly polymorphic (Supplementary material 1). Nevertheless, these polymorphisms were particular of each taxon not reducing the phylogenetic information of ‘host order’ as shown by the CIs (Table 2). The same patterns, in a less intense way, were observed for the character ‘biogeographic occurrence’ (Table 2; Supplementary material 1).
The remaining characters used in the concatenated data analysis showed random mapping in the phylogenies, appearing independently in some terminal lineages and showing paraphyletic patterns. The CIs of these characters were generally low, except by the ‘relative position of excretory pore’ and ‘number of caudal spikes on tail in males’ in the 28S dataset, as well as ‘vulval lips protrusion’ and ‘general morphology of tail in females’ in the 28S and COI datasets (CIs ⩾0.667, see Table 2).
Discussion
The newly collected camallanids from A. endy and C. micracanthus, used for genetic characterization, showed identical morphology as in previous taxonomic descriptions of S. hilarii and S. pintoi, respectively, including those of recent reports in closely related hosts from Argentina (see Moravec, Reference Moravec1998; Ramallo and Ailán-Choke, Reference Ramallo and Ailán-Choke2017; Ailán-Choke et al., Reference Ailán-Choke, Ramallo and Davies2018). Spirocamallanus pintoi has been previously reported in C. micracanthus from Argentina (Ailán-Choke et al., Reference Ailán-Choke, Ramallo and Davies2018); and its 18S genetic similarity with a conspecific sequence in GenBank, reinforced the present morphological identification. Moreover, fragments of the 28S and COI were sequenced for the first time for S. pintoi. Currently, there is no genetic data available for S. hilarii preventing comparison and, consequently making the present results, the first genetic characterization for the species. Since both species have no taxonomic problems and their morphology is well-known, giving morphological data here is not necessary.
During the preliminary searches, it was possible to observe problematic and fragmented data in both genetic and morphological databases related to camallanids. Some species descriptions were poorly detailed, mainly with respect to the cephalic structures, relative position of deirids and excretory pore (e.g. in Ivashkin et al., Reference Ivashkin, Sobolev and Khromova1971; Stromberg et al., Reference Stromberg, Shegog and Crites1973; Pinto et al., Reference Pinto, Fábio, Noronha and Rolas1974). In the genetic dataset, we also found problematic data as reported by previous studies (Wijová et al., Reference Wijová, Moravec, Horák and Lukeš2006; Černotíková et al., Reference Černotíková, Horák and Moravec2011; Ailán-Choke et al., Reference Ailán-Choke, Davies, Tavares and Pereira2019, Reference Ailán-Choke, Tavares, Luque and Pereira2020). In this sense, representatives of C. cotti, C. oxycephalus and S. istiblenni from India were not considered here, based on their anomalous phylogenetic behaviour, which suggests their misidentification (see Ailán-Choke et al., Reference Ailán-Choke, Tavares, Luque and Pereira2020). Similarly, one of two 18S sequences available for S. rarus, isolated from catfish in Peru, is known as misidentified (Černotíková et al., Reference Černotíková, Horák and Moravec2011; Ailán-Choke et al., Reference Ailán-Choke, Davies, Tavares and Pereira2019, Reference Ailán-Choke, Tavares, Luque and Pereira2020); therefore, we considered the one (JF803912) closest to representatives parasitizing Siluriformes from the Neotropical region to be the most accurate, based on the present and previous phylogenies (Ailán-Choke et al., Reference Ailán-Choke, Tavares, Luque and Pereira2020). The presence of inaccurate and lacunar data reported here, still represent a real challenge when dealing with the phylogeny and taxonomy of Camallanidae (Wijová et al., Reference Wijová, Moravec, Horák and Lukeš2006; Černotíková et al., Reference Černotíková, Horák and Moravec2011; Ailán-Choke et al., Reference Ailán-Choke, Davies, Tavares and Pereira2019, Reference Ailán-Choke, Tavares, Luque and Pereira2020).
Analysing the phylogenetic informativeness of genetic markers a continuous scale was observed, in which the nuclear 18S and the mitochondrial COI occupied the extremes, showing peaks of information among deeper and shallower nodes, respectively. Large mutational rate and, consequently, higher phylogenetic informativeness among lower taxa (i.e. species level) is expected to be found comparing mitochondrial vs nuclear markers in invertebrates (Allio et al., Reference Allio, Donega, Galtier and Nabholz2017; Kern et al., Reference Kern, Kim and Park2020). The unexpected high rate of genetic variation across 28S sequences observed here was similar to previous reports (Kuzmin et al., Reference Kuzmin, Tkach, Snyder and Bell2011; Svitin et al., Reference Svitin, Truter, Kudlai, Smit and du Preez2019) and seems to be common in Camallanidae, mainly in the region D2 of the gene. Obviously, it would be better to concatenate the datasets in order to maximize the genetic information; however, the scarcity of taxa correspondence between these datasets would dramatically reduce the number of representatives, making the analysis meaningless.
Nodal supports in the phylogenetic hypotheses inferred from genetic data followed the pattern observed in the phylogenetic informativeness analysis, in which most of the deeper nodes were weakly supported in COI analysis. The lowest CIs of trees were observed in this dataset, where trees inferred from genetic and concatenated data showed statistically lower CIs than that of the tree inferred from morphological traits. These results indicate that the present morphological data (mainly some traits of buccal capsule and morphology of tail in males) were more consistent with the nuclear markers than with the mitochondrial.
As previously mentioned, the overall information of the phylogenetic hypotheses from genetic and concatenated trees were quite similar, except for in the analyses including COI. In this case, the presence of morphological data brought greater stability to the lineages, improving nodal supports. A similar stability, but without support improvement could be observed in the trees of 18S and 28S datasets. These trends reinforce the importance of integrative approaches on the taxonomy of complicated taxa such as Camallanidae and, even though the present analysis was limited by data availability, some interesting insights could be noted and are discussed as follows.
According to the conception of Moravec and Thatcher (Reference Moravec and Thatcher1997), the genus Procamallanus includes the subgenera Procamallanus and Spirocamallanus. This classification was not supported by the present analysis and should not be adopted. Therefore, the generic status of Procamallanus and Spirocamallanus was considered valid here, following the conception of Petter (Reference Petter1979).
The genus Batrachocamallanus was erected by Jackson and Tinsley (Reference Jackson and Tinsley1995) to accommodate the species of Procamallanus with small-sized females, bearing more than three spikes (mucrons) on tail end and parasitizing amphibians; its validity has been accepted by some authors (Svitin et al., Reference Svitin, Schoeman and du Preez2018, Reference Svitin, Truter, Kudlai, Smit and du Preez2019) and rejected by others (Moravec et al., Reference Moravec, Justine, Würtz, Taraschewski and Sasal2006). The present results indicated close relatedness between Batrachocamallanus and species of Procamallanus. Moreover, based on character mapping, the state ‘more than 3 spikes (mucrons) on female tail’ and ‘amphibian host order’, which are diagnostic for Batrachocamallanus, appeared independently in lineages of Camallanus (e.g. C. lacustris, C. kaapstaadi and C. xenopodis) and Procamallanus (i.e. P. laeviconchus, P. pseudolaeviconchus and P. pacificus), as well as in S. fulvidraconis and Se. octorugatum (Baylis, 1933). These results and the polyphyly of Procamallanus and Spirocamallanus suggest no current support validating Batrachocamallanus and, following Moravec et al. (Reference Moravec, Justine, Würtz, Taraschewski and Sasal2006), species of this genus should reallocated as follows: P. silurinae (Jackson & Tinsley, Reference Jackson and Tinsley1995), P. slomei (Southwell & Kirshner, 1937), S. occidentalis (Jackson & Tinsley, Reference Jackson and Tinsley1995) and S. xenopodis (Baylis, 1929). It should be mentioned that Svitin et al. (Reference Svitin, Truter, Kudlai, Smit and du Preez2019) accepted the validity of Batrachocamallanus based on genetic criteria, but the referred dataset was reduced and the polyphyly of Procamallanus were not evidenced (Procamallanus and Spirocamallanus were represented by one species each). As long as there is no strong set of characters or an autapomorphy that defines Batrachocamallanus, and due to its close relatedness with the polyphyletic genera Procamallanus and Spirocamallanus, this genus should not be validated (Moravec et al., Reference Moravec, Justine, Würtz, Taraschewski and Sasal2006).
Bringing morphological characters to an integrative approach can be a ‘double-edged sword’ and their roles should be evaluated with caution. The buccal capsule traits showed high CI values, indicating low degree of homoplasy, and were important to confirm the close relatedness of congeners of Camallanus, as well as between this genus, Paracamallanus and Serpinema (i.e. presence of scletorized tridents, longitudinal ridges and the articulation of into valves). Similarly, the presence of conspicuous-bladed teeth at the buccal capsule base was consistent with the fully supported clade formed by S. huacraensis and S. rarus. However, the buccal capsule structure was not consistent enough to support the monophyly of Camallanus, Procamallanus and Spirocamallanus. Therefore, the artificiality of the current systematics of Camallanidae, mainly based on the buccal capsule morphology for generic/subgeneric diagnosis, is confirmed (Wijová et al., Reference Wijová, Moravec, Horák and Lukeš2006; Černotíková et al., Reference Černotíková, Horák and Moravec2011; Ailán-Choke et al., Reference Ailán-Choke, Davies, Tavares and Pereira2019, Reference Ailán-Choke, Tavares, Luque and Pereira2020).
The states of morphological characters present on the tail of males, including presence/absence of caudal alae, pedunculate papillae and spicule symmetry were highly consistent with each other. It may be possible that the presence of caudal alae, pedunculate papillae and asymmetric spicules represent ancestral states, been lost (or modified) during different times and events (as indicated by character mapping), in agreement with the traditional systematics by Chabaud and Bain (Reference Chabaud and Bain1994). A similar pattern may be related to the presence of gubernaculum, which is associated with the group of species having caudal alae; this structure seems to appear independently along the time, but in more recent lineages. These trends were better observed in the phylogenies of 18S dataset, as a result of higher number and diversity of taxa included.
According to Petter (Reference Petter1979), species of Spirocamallanus with caudal alae, pedunculate papillae and asymmetric spicules, are mostly parasites of marine fishes. Petter (Reference Petter1979) and Rigby and Adamson (Reference Rigby and Adamson1997) pointed out that the morphology of tail in females is likewise constant within a species of Spirocamallanus. These parameters, along with the structure of cephalic end, the relative position of deirids, excretory pore, female tail structure and vulva, may be adequate for interspecific comparisons, but proved to be polymorphic on the higher systematics of Camallanidae. The high CIs observed for these characters in concatenated trees of 28S and COI analyses, along with CIs of ‘number of caudal spikes on tail in males’ and ‘vulval lips protrusion’, were biased by the low representativeness of taxa. Even though the relative position of deirids appeared to be consistent with some assemblages in the 18S dataset (with CI > 0.6 in the concatenated data tree), we believe that the result was circumstantial and other factors have greater consistency with the phylogenetic configuration of Camallanidae (see below).
In most of the lineages from freshwater hosts, males showed a similar caudal morphology; the same was observed in species from marine hosts. In addition to that, the results indicated that ‘habitat’ is an important factor on the phylogeny of camallanids (see also CI values in Table 2). Unfortunately, only the 18S dataset included more than one sample from the marine environment, which formed a moderate and a strong supported lineage in the genetic and concatenated data trees, respectively (except for by S. rebecae that occurs in freshwater). Therefore, the phylogenetic sense of male caudal morphology and habitat are more closely related to genetic data, than most of the characters evaluated here. These results suggest a likely marine origin of camallanids as early asserted by Petter (Reference Petter1979) and Chabaud and Bain (Reference Chabaud and Bain1994), based on morphological and life history traits of parasites (see the two previous paragraphs). This fact does not preclude the reversal from freshwater to marine habitat in more recent lineages as observed in Camallanus spp.
Similarities in ‘host order’ were observed in several closely related lineages from different trees and datasets, for example, in assemblages of Camallanus parasitic in turtles and in those of Procamallanus and Spirocamallanus parasitic in Perciformes and Siluriformes. Therefore, host taxa and, consequently, the habitat (as previously discussed) are strong factors influencing the evolutionary paths within Camallanidae. In this sense, some host–parasite evolutionary patterns may be related, as observed among the species of Procamallanus and Spirocamallanus parasitic in Characiformes, Siluriformes and/or Anguiliformes that were closer to each other, than to those parasitic in Perciformes. These patterns reflect the pattern of relatedness observed within Actinopterygii (see Betancur et al., Reference Betancur, Wiley, Arratia, Acero, Bailly, Miya, Lecointre and Ortí2017; Hughes et al., Reference Hughes, Orti, Huang, Sun, Baldwin, Thompson, Arcila, Li, Becker, Bellora, Zhao, Li, Wang, Fang, Xie, Zhou, Huang, Chen, Venkatesh and Shi2018). In parallel, some recent lineages became parasites of amphibians and reptiles, which may have happened by host capture as proposed early by Chabaud and Brygoo (Reference Chabaud and Brygoo1962). These are all plausible ideas illustrated by the present results and suggested in previous studies (Chabaud and Brygoo, Reference Chabaud and Brygoo1962; Petter, Reference Petter1979; Chabaud and Bain, Reference Chabaud and Bain1994); however, much effort is needed prior to the achievement of more solid conclusions.
It seems that the biogeography agrees with the same patterns observed for habitat and host order, but similarities on the states of ‘biogeographic origin’ were less strongly related to the lineages of the phylogenetic hypotheses. Freshwater species from the Neotropical, Afrotropical and Australasian regions were close to each other. Similarly, marine species from Pacific were closer to each other than with those from Atlantic. Thus, it is undeniable (and expected) that the parasite biogeography is linked with host and habitat characteristics, and also influences the phylogenetic configuration of Camallanidae (Chabaud and Bain, Reference Chabaud and Bain1994). It should be commented that the degree of influence is different depending on the genera of camallanid. Non-linearity in the influence of host, habitat and biogeographic features in evolutionary assemblages of parasites is common (Rohde, Reference Rohde2002; Blasco-Costa et al., Reference Blasco-Costa, Rouco and Poulin2015; Pérez-Ponce de León and Choudhury, Reference Pérez-Ponce de León and Choudhury2015).
The only polymorphic states, observed in few species, were related to ‘host order’ and ‘geographic origin’, indicating their wide host and geographical ranges. However, it did not bias the analysis of these characters, since the polymorphic profile was particular to each taxon. In the phylogenies, only the host and geographic characteristics intrinsic to the isolate used for genetic sequencing were depicted, in order to highlight the fidelity between genetic and concatenated data trees, since host taxa may be related to the parasite genotype (see Ailán-Choke et al., Reference Ailán-Choke, Tavares, Luque and Pereira2020). The CI values obtained for ‘host order’ and ‘geographic origin’ reinforced their phylogenetic consistency, despite the polymorphisms.
The only monophyletic assemblage was the clade formed by Camallanus, Paracamallanus and Serpinema. Even though the monophyly of Camallanus was not fully supported due to Serpinema, Camallanus spp. showed a strong tendency to cluster together in the same clade, partially overshadowed the phylogenetic similarities on habitat, host taxa and biogeography. Conversely, the evolutionary paths of Procamallanus and Spirocamallanus diverged independently during different times, with different ancestry patterns, but the assemblages showed similarities on habitat, host taxa and biogeography. Unfortunately, the limited number of representatives of Paracamallanus and Serpinema prevents us from discussing their cladistics. Nevertheless, the results suggested that ancestral lineages of Camallanidae are closer to Procamallanus and Spirocamallanus than Camallanus, Paracamallanus and Serpinema.
Important phylogenetic patterns involving genetic, morphological and life history correspondence were observed here, mainly with respect to the caudal morphology of males, habitat, host taxa, biogeography and finally some aspects of the buccal capsule structure. Habitat, host taxa and biogeography are closely linked to each other and with the phylogeny of Camallanidae. These similarities exhibit different degrees of association, varying according to each generic taxon. Nevertheless, the origin of camallanids most likely occurred in marine fishes. The current systematics of Camallanidae mainly based on buccal capsule is artificial, but shows some consistency with the classification of Camallanus, Paracamallanus and Serpinema. However, it does not support subgenera and, consequently, Procamallanus and Spirocamallanus should be considered valid independent genera. Most of the morphological and life history traits covered here, especially the general morphology of buccal capsule and male tail, habitat, host taxa and biogeographic patterns, are adequate in grouping for species comparison and diagnosis, because they are particularly consistent with closely related terminal lineages. Explanations for the few exceptions to the various patterns observed here will be elucidated as new sequences from different regions of the genome are added to the database. The present results largely agree with previous findings (Wijová et al., Reference Wijová, Moravec, Horák and Lukeš2006; Černotíková et al., Reference Černotíková, Horák and Moravec2011; Sardella et al., Reference Sardella, Pereira and Luque2017; Ailán-Choke et al., Reference Ailán-Choke, Davies, Tavares and Pereira2019, Reference Ailán-Choke, Tavares, Luque and Pereira2020), and even with the difficulties of limited and inaccurate data, important results that will help clarifying the phylogeny of Camallanidae were achieved.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182021000706
Acknowledgements
The authors thank Dr Luiz Eduardo Roland Tavares from the Universidade Federal de Mato Grosso do Sul and Dr Carina Elisei from the Universidade Católica Dom Bosco for providing the inputs and facilities for molecular procedures. Additionally, the authors thank Dr František Moravec, Dr Tomás Scholz and Blank Škoríková from the Institute of Parasitology of the Czech Academy of Sciences, in České Budějovice, for providing important scientific literature. Thanks are also due to Dr Dora Davies, Florencia Liquin, Jose Saravia and Roberto Sanchez for their assistance in field collections and laboratorial procedures.
Author contributions
LGA-C and FBP conceived and designed the study, conducted data gathering, performed the analyses and wrote the article.
Financial support
This study was financed in part by Consejo de Investigación, Universidad Nacional de Salta, Argentina (grant number 2481/0). LGA-C was supported by a Post-doctoral fellowship from the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET, Argentina).
Conflict of interest
The authors declare there are no conflicts of interest.
Ethical standards
All procedures involving animal manipulation were approved by the Secretaría de Medio Ambiente, Ministerio de Ambiente y Producción Sustentable, Gobierno de la provincia de Salta, Argentina (Authorization No. 000248/14), strictly according to the international ethical standards in animal research.