INTRODUCTION
The exponential decay of similarity in species composition between two communities as a function of the distance separating them has emerged as a more-or-less universal phenomenon in biogeography (Nekola and White, Reference Nekola and White1999; Soininen et al. Reference Soininen, McDonald and Hillebrand2007; Morlon et al. Reference Morlon, Chuyong, Condit, Hubbell, Kenfack, Thomas, Valencia and Green2008). For example, in communities of parasitic organisms, pairwise similarity decreases with increasing geographical distance between host populations sampled, whether these involve fish (Poulin, Reference Poulin2003; Oliva and González, Reference Oliva and González2005; Seifertova et al. Reference Seifertova, Vyskocilova, Morand and Simkova2008; Pérez-del-Olmo et al. Reference Pérez-del-Olmo, Fernández, Raga, Kostadinova and Morand2009), mammals (Poulin, Reference Poulin2003; Brouat and Duplantier, Reference Brouat and Duplantier2007; Vinarski et al. Reference Vinarski, Korallo, Krasnov, Shenbrot and Poulin2007), or invertebrates (Thieltges et al. Reference Thieltges, Ferguson, Jones, Krakau, de Montaudouin, Noble, Reise and Poulin2009). Processes similar to those responsible for generating this common pattern should apply to scales other than spatial distance, however. Thus the exponential decay of similarity should be manifested along any dimension characterizing some type of separation between communities.
In the case of parasite communities, some of the parasite species exploiting two closely-related host species will either be the same on both hosts or they will be close relatives, having been inherited by descent from a common ancestor of the two hosts (Brooks, Reference Brooks1988; Brooks and McLennan, Reference Brooks and McLennan1993). The remaining parasite species, not passed down from an ancestral host, may also include shared species acquired more recently, but these should differ in proportion to the differences existing between the two host species with respect to ecology, physiology and immunology. These differences are best captured by the phylogenetic affinities between host species. Thus, regardless of how parasite species came to exploit particular hosts, phylogenetic distance between host species, measured as the relative number of base-pair differences along homologous DNA sequences, could be another dimension along which similarity in parasite species composition should decrease exponentially. Two of the key processes shaping distance decay in geographical space are autocorrelation of environmental variables and dispersal constraints (Steinitz et al. Reference Steinitz, Heller, Tsoar, Rotem and Kadmon2006; Soininen et al. Reference Soininen, McDonald and Hillebrand2007). First, climate and other abiotic conditions often follow parallel gradients, with two nearby localities more likely to show similar conditions than two distant locations. Since these conditions determine which species can establish and maintain populations in those localities, nearby sites with identical conditions are likely to support similar communities. Similarly, closely-related host species have more characteristics in common than distantly-related species; this obvious fact forms the basis of comparative methods in which phylogenetic influences must be taken into account when comparing species (Felsenstein, Reference Felsenstein1985; Harvey and Pagel, Reference Harvey and Pagel1991). Closely-related host species, because they offer similar living conditions for parasites, use similar immune defences against parasites, and experience similar risks of acquiring the same parasite species, should therefore have more similar parasite faunas than unrelated host species. Second, in geographical space, a species has a greater probability of dispersing to a nearby locality than to a distant one, resulting in a decrease in community similarity with increasing distance. The same happens in phylogenetic space: host-switching, i.e. the colonization of a new host species by a parasite, is more likely if the new host is related to, and sympatric with, the original one (Brooks, Reference Brooks1988): the greater the phylogenetic distance between two host species, the greater the ‘jump’ for the parasite. The upshot is that related host species should harbour more similar parasite faunas than unrelated ones.
The influence of host phylogeny on the similarity in parasite species richness between host species has been highlighted by comparative studies. Typically, a significant relationship between parasite species richness and any given host trait becomes non-significant, or at least weaker, when the analysis is repeated while correcting for phylogenetic effects (Gregory, Reference Gregory1990; Poulin, Reference Poulin1995, Reference Poulin1997; Nunn et al. Reference Nunn, Altizer, Jones and Sechrest2003). This indicates that how many, and most likely which, parasite species are harboured by a host species are not properties independent of its phylogenetic affinities. What has not been quantified to date, however, is how similarity in the composition of the parasite faunas of different host species decreases with increasing phylogenetic distance. If the pattern observed almost universally in geographical space extends to phylogenetic space, we would expect an exponential decay relationship, or, in a log-log plot, a linear function between these two variables. The ability of parasites to disperse in phylogenetic space, i.e. to colonize new host species, should impact the rate of decay. Some parasite taxa are notoriously host-specific, i.e. restricted to only one or very few related host species (Poulin, Reference Poulin1992; Poulin and Mouillot, Reference Poulin and Mouillot2003; Poulin and Keeney, Reference Poulin and Keeney2008). This is true, for instance, of ectoparasitic taxa such as monogeneans or lice. These parasite groups have generally tended to co-speciate closely with their hosts over evolutionary time, but they may in some cases show high rates of host-switching if they have a dispersal route (Hafner and Nadler, Reference Hafner and Nadler1988; Desdevises et al. Reference Desdevises, Morand, Jousson and Legendre2002; Clayton and Johnson, Reference Clayton and Johnson2003; Huyse and Volckaert, Reference Huyse and Volckaert2005). Among extant parasite assemblages, a parasite species pool consisting mostly of host-specific taxa should show steeper decay of similarity as a function of phylogenetic distance than one comprising mostly generalists. Therefore, both the shape and the slope of the decay relationship can shed light on the parallels between biogeographical processes and evolutionary ones.
Here, I investigate the decrease in similarity of parasite faunas with increasing phylogenetic distance among host species, for the metazoan parasites in 5 families of Canadian freshwater fishes. The parasite faunas of Canadian fishes have been the subject of several hundred scientific investigations over the past many decades, resulting in comprehensive compilations of the parasites found in each fish species (Margolis and Arthur, Reference Margolis and Arthur1979; McDonald and Margolis, Reference McDonald and Margolis1995). As a whole, the parasites of Canadian fishes represent one of the (if not the) best-known assemblages of parasites in vertebrates of any geographical region (Poulin, Reference Poulin2004). The specific objectives of this study are (i) to determine the shape of the function linking similarity and phylogenetic distance across all fish as well as in each of the 5 host families, and (ii) to determine whether the rate of decay in similarity of parasite faunas differs among different taxonomic groups of parasites. In other words, I will test whether the decay relationship is universal or whether it depends on the type of hosts or parasites considered.
MATERIALS AND METHODS
Data on the metazoan parasite fauna of fish species in the families Percidae, Centrarchidae, Ictaluridae, Cyprinidae, and Catostomidae were included in the analyses. Only fish categorized as freshwater species and with at least 25% of their geographical range within Canada were included (see Scott and Crossman, Reference Scott and Crossman1973); this ruled out a few fish species, in particular cyprinids. Also excluded were fish species with fewer than 3 parasite genera exploiting them, or fish species for which no phylogenetic information was available (see below). For each fish species, lists of known metazoan parasite species were compiled from Margolis and Arthur (Reference Margolis and Arthur1979) and McDonald and Margolis (Reference McDonald and Margolis1995). These included only the taxa that form long-term associations with fish, i.e. monogeneans, trematodes, cestodes, nematodes, acanthocephalans, and copepods. Thus, leeches and branchiurans were not included. Since synonymies or errors in parasite identification may affect similarity measures, and since some entries only specified the genus of the parasite and not its species, all analyses were performed at the more conservative level of the parasite genus. Therefore, for each fish species, the presence or absence of each parasite genus was entered in a host-parasite association matrix.
For each fish family and their parasites, 2 additional variables were also recorded: (i) the host specificity of each parasite genus, i.e. the number of host species in the host family from which it has been found, and (ii) the parasite generic richness of each fish species, i.e. the number of parasite genera infecting that fish. In addition, for each subset of host and parasite species analysed, 2 other variables were noted, both of which may act as confounding factors in the analyses: (iii) the size of the host-parasite matrix, equal to the number of host species multiplied by the total number of parasite genera, and (iv) the matrix fill, or the number of realised host-parasite combinations in the species-by-genera matrix expressed as a percentage of the total number of possible combinations. This last measure is obviously related to the first two: if all parasite genera infect most host species in a family, or if all host species harbour most of the parasite genera in the available pool, then matrix fill will be high.
Within each fish family, the similarity in the parasite faunas of 2 fish species was computed for all possible pairs of host species, using the Jaccard index, one of the most widely used indices of ecological similarity (Magurran, Reference Magurran1988). It corresponds to the number of parasite genera shared by 2 host species divided by the total number of parasite genera occurring in the 2 host species put together; it ranges from zero (no parasite in common between the 2 host species) to 1 (the 2 host species have exactly the same parasites). Pairwise similarities between fish species were first computed including all parasite taxa, and then separately for each of the 4 taxa represented by enough genera: monogeneans, trematodes, cestodes and nematodes. All similarities were computed using EstimateS (Colwell, Reference Colwell2006).
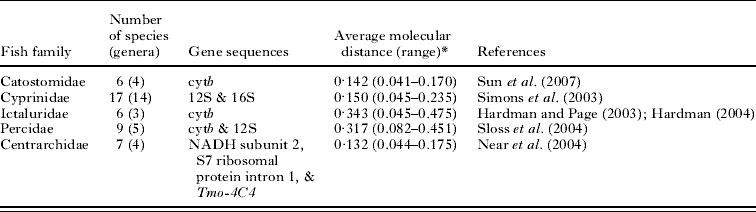
Molecular phylogenies were used to obtain all pairwise phylogenetic distances between fish species within each of the 5 families. The phylogenies used were based on nuclear and (mostly) mitochondrial DNA sequences and maximum likelihood analyses; when sequence data for more than 1 gene were used in the original reconstruction, the phylogeny resulting from the combined data was used (Table 1). Based on genetic distances measured as base-pair differences obtained from each phylogenetic tree, the total genetic divergence, or phylogenetic distance, between all pairs of species was calculated in units of number of substitutions per site (Table 1).
Table 1. Summary of the data used to estimate pairwise phylogenetic distances among fish species in five families

* Proportion of base-pair differences (i.e. substitutions per site).
Phylogenetic distance values and Jaccard similarity values were log10-transformed (or log10[x+1]-transformed in subsets with Jaccard values of zero). Linear regressions in log-log space were then performed on these data, separately for each fish family. As the data are not truly independent in a statistical sense (each host species is used in more than 1 pairwise comparison), and to account for slight deviations from normality in data distribution, the significance of each regression was validated using a randomization approach (Manly, Reference Manly1997), implemented using the RT 2.1 software (Western EcoSystems Technology Inc., Cheyenne, WY, USA); all regression P-values reported are therefore the more conservative ones based on 1000 permutations. These regressions were initially computed separately for each fish family. Subsequently, to search for general patterns across all fish species, 2 approaches were used. First, a linear regression was performed as above but after pooling all pairwise comparisons from all 5 fish families together. Second, a meta-analysis was conducted on the results obtained for separate fish families. Effect sizes were computed for each family as Fisher z-transformation of the original correlation coefficients between log-transformed phylogenetic distance and Jaccard similarity values, with the number of pairwise comparisons as sample size. A combined estimate of the overall effect size was computed using the software package Comprehensive Meta-Analysis 2.0 (Biostat, Englewood, NJ, USA).
Finally, to test whether data points in the significant regressions were more concentrated in a triangular area of the bivariate space than expected by chance, a boundary test was performed using the EcoSim 7 software (Gotelli and Entsminger, Reference Gotelli and Entsminger2004). After defining a square data space delimited by the minimum and maximum values of both variables, and splitting this space with a diagonal into an upper right triangle and a lower left triangle, this test uses a null-model approach to calculate the number of points that fall on either side of the diagonal (boundary) and their sum of squares, for observed and simulated data (1000 randomizations, asymmetric distribution option). If, for instance, the lower left corner of the space is unusually empty of points, the number of points in that corner and/or their sum of squares in the observed data set will be significantly lower than in the simulated data sets.
RESULTS
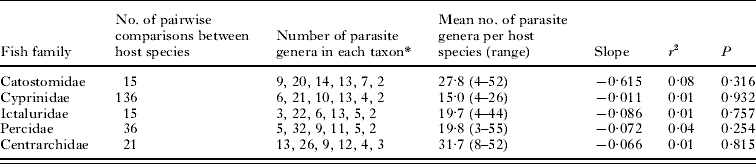
Data were obtained for 45 fish species belonging to 30 genera and 5 families. The parasite genera exploiting those fish hosts show a certain degree of host specificity (Fig. 1). The majority have been recorded from no more than 4 fish species within a family, though a very few can infect all host species within the family. The numbers of parasite genera harboured per fish species also varied widely, with some fish hosting only 3–4 parasite genera and others known to serve as host for more than 50 (see Table 2).

Fig. 1. Frequency distributions of host specificity, i.e. number of host species exploited, among the parasite genera infecting 5 families of Canadian freshwater fishes.
Table 2. Descriptive data and regression statistics (in log-log space) for the decay of similarity with phylogenetic distance among the parasite assemblages of fish host species
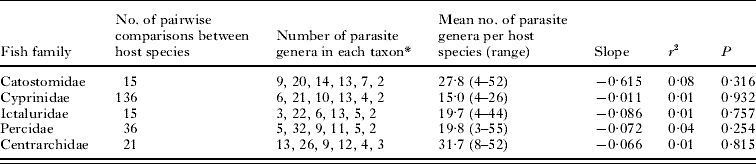
* In order: monogeneans, trematodes, cestodes, nematodes, acanthocephalans, and copepods.
Jaccard similarity values indicate that, on average, 2 fish species belonging to the same genus share 36% of their parasite genera, whereas 2 fish from different genera have only 29% of their parasite genera in common. However, these values vary among fish families, and are based on very few within-genus comparisons (Fig. 2). In particular, similarity in the composition of the parasite fauna shows no sign of being greater on average between congeneric fish species than between fish from different genera within the Percidae and Ictaluridae (Fig. 2). Nevertheless, when considering all comparisons from all 5 fish families together, there is a general decrease in similarity of the parasite fauna as a function of increasing phylogenetic distance between host species (Fig. 3). The significant linear function (r 2=0·07, P=0·008) in log-log space suggests an exponential decay in similarity with increasing phylogenetic distance. However, phylogenetic distance explains only a small proportion of the variance in similarity values, and the slope of the linear function (−0·332) has a broad 95% confidence interval (−0·495 to −0·054). The distribution of data points in the scatterplot was significantly aggregated in the upper right triangle of the bivariate space (Fig. 3). Both the boundary test based on the number of points (P=0·038) and that based on sums of squares (P=0·021) confirmed this. This suggests that 2 phylogenetically close host species tend to share a high proportion of parasite genera, but that phylogenetically distant host species have roughly equal chances of harbouring very similar or very dissimilar parasite faunas.

Fig. 2. Mean (±s.e.) Jaccard index of similarity, i.e. proportion of shared parasite genera, in pairwise comparisons between host species from the same genus and between host species from different genera, for 5 families of Canadian freshwater fishes. The number of comparisons involved is given above each bar.

Fig. 3. Distance decay of similarity (Jaccard index) among the parasite assemblages of different host species as a function of increasing phylogenetic distance between host species, in log-log space. Phylogenetic distance is expressed as the proportion of base pair differences (i.e. substitutions per site) in homologous DNA sequences. Pairwise comparisons within different fish families are indicated by different symbols.
These analyses were then performed separately for monogeneans, trematodes, cestodes and nematodes. For 2 groups of parasites, the same significant decay of similarity with increasing phylogenetic distance among host species was observed (monogeneans: slope=−0·080, r 2=0·03, P=0·034; trematodes: slope=−0·237, r 2=0·04, P=0·023). In these analyses too, data points were significantly aggregated in the upper right corner of the scatterplots (boundary tests, all P<0·045). However, because of wide 95% confidence intervals, the slope values obtained for monogeneans and trematodes did not differ from each other, or from that obtained for all parasite groups combined. In contrast, there was no significant decay of similarity with increasing phylogenetic distance in either cestodes (slope=0·009, r 2=0·001, P=0·691) or nematodes (slope=−0·045, r 2=0·014, P=0·077).
The different fish families considered here are not characterized by highly similar values for phylogenetic distances between pairs of species (e.g. contrast points for Cyprinidae and Percidae in Fig. 3; see also Table 1). When the above analyses were performed separately for each fish family, and including all parasites, no significant decrease in similarity of the parasite fauna as a function of phylogenetic distance between hosts was observed, although all slope values were negative (Table 2). Based on the meta-analysis, the combined effect size was weak and not quite significant (correlation: −0·125, 95% confidence interval −0·256 to 0·101; z-value=−1·81, P=0·070). Similarly, analyses performed within each host family and separately for either only monogeneans, trematodes, cestodes or nematodes all yielded non-significant relationships (not shown, all regressions P>0·30; meta-analyses, all P>0·10).
The full set and the different subsets of hosts and parasites considered above include different numbers of taxa, and are all characterized by different mean host specificity (for the parasite genera involved) and mean parasite generic richness (for the host species). The analyses that yielded a significant decay of similarity are those including all fish species together, and they are thus those with the largest host-parasite matrices. Matrix fill, which reflects both host specificity and mean parasite generic richness, could be a confounding factor, if, for instance, a high proportion of zeros in the matrix reduces the probability of detecting significant decay. However, a comparison of matrix fill values between analyses that produced a significant result and those that did not failed to find a significant difference (t-test, P=0·34).
DISCUSSION
Because parasites and their hosts are often associated by descent, many parasite species on extant host species have simply been inherited from ancestral hosts (Brooks, Reference Brooks1988; Hafner and Nadler, Reference Hafner and Nadler1988; Clayton and Johnson, Reference Clayton and Johnson2003). This should inevitably lead to phylogenetically closely related host species sharing a few, if not several, parasite genera. The fact that these parasites have not been acquired independently justifies the need to control for phylogenetic relatedness in comparative analyses linking host life-history traits to parasite diversity (Poulin, Reference Poulin1995; Nunn et al. Reference Nunn, Altizer, Jones and Sechrest2003). The assumption that related host species have some parasite species in common because of their common origin is qualitatively sound, but has never been properly assessed quantitatively. The present study is the first attempt to quantify the relationship between phylogenetic relatedness and the proportion of shared parasite taxa. Its results indicate that similarity in parasite faunas tends to decay exponentially, but not strongly, with increasing phylogenetic divergence between host species, a finding that parallels those of studies investigating community similarity among localities as a function of geographical distance (Nekola and White, Reference Nekola and White1999; Poulin, Reference Poulin2003; Soininen et al. Reference Soininen, McDonald and Hillebrand2007; Morlon et al. Reference Morlon, Chuyong, Condit, Hubbell, Kenfack, Thomas, Valencia and Green2008).
However, the decrease in similarity of parasite faunas as a function of host phylogenetic distance seen in the present study appears relatively weak for at least 4 reasons. First, in quantitative terms, the decay of similarity is not pronounced. For example, compared to 2 fish species separated genetically by 0·1 substitution per site, 2 fish that are 3 times more phylogenetically distant, i.e. separated by 0·3 substitutions per site, would only differ by a further 9% of their parasite fauna (based on the regression slope). Second, the significant clustering of points in the upper right corner of the scatterplot indicates that many pairs of fish species that are phylogenetically distant can still share a high proportion of parasites. Several pairs of percid fishes, such as the yellow perch Perca flavescens and the darter Etheostoma nigrum, provide examples of this pattern. Third, the significant regressions indicate that phylogenetic distance only explained a small percentage of the total variance in similarity between host species. Fourth, a significant decay with distance was only observed when pairwise comparisons from all 5 fish families were pooled, and never when they are treated separately. This last finding is supported by the meta-analysis, which showed no significant combined effect among within-family analyses.
All this suggests that although phylogenetic affinities may increase the likelihood of parasite sharing, other factors also play important roles. Similar ecological attributes, such as microhabitat use or diet, can independently expose unrelated fish species to colonization by the same parasite taxa. In the Centrarchidae, ontogenetic changes in diet mean that even the largest species from the genus Micropterus overlap in diet, at least early in their life before they become piscivorous, with smaller species in the genera Pomoxis and Lepomis (Scott and Crossman, Reference Scott and Crossman1973). Therefore most centrarchid species, regardless of their phylogenetic affinities, include the same range of invertebrate prey in their diet, and can all potentially acquire the same helminth parasites using these invertebrates as intermediate hosts. Similarly, in the Percidae, young of the large-bodied species in the genus Stizostedion have a similar diet to that of the small-bodied members of the darter genera Etheostoma and Ammocrypta (Scott and Crossman, Reference Scott and Crossman1973). Also, an analysis of the origins of host-parasite associations in the yellow perch, Perca flavescens, has shown that this fish has acquired the vast majority of its parasites via host-switching from sympatric fish species with similar feeding habits, whether these are percids or not (Carney and Dick, Reference Carney and Dick2000). Phylogenetic distances based on differences in DNA sequences do not reflect perfectly the true ecological or physiological distances between host species, in terms of exposure and susceptibility to parasite invasion. Two host species that are phylogenetically very close must inevitably share many parasite taxa, whereas two host species that are not particularly close may or may not share many parasites, depending on the degree of ecological divergence between them. This would explain the generally triangular scatter of points seen in all significant regressions between parasite similarity and host phylogenetic distance.
A significant decay of similarity with increasing host phylogenetic distance was observed separately for monogenean and trematode parasites, but not for cestodes or nematodes. There may be intrinsic biological differences between these groups of parasites that limit the probability of host-switching across relatively large host phylogenetic gaps in monogeneans and trematodes, but not in cestodes or nematodes. Monogeneans are notoriously highly host specific, with many species being restricted to a single host species (Lambert and El Gharbi, Reference Lambert and El Gharbi1995). Among parasites of Canadian freshwater fish, monogeneans and, to a lesser extent, adult trematodes are on the whole more host specific than adult cestodes and nematodes (Poulin, Reference Poulin1992). In contrast, larval trematodes (i.e. metacercarial stages), which use fish as intermediate hosts, are generalists that show little host specificity compared to adult stages, but the same is true of larval nematodes and cestodes (Margolis and Arthur, Reference Margolis and Arthur1979; McDonald and Margolis, Reference McDonald and Margolis1995). Among the taxa considered here, there was overall a greater proportion of trematodes occurring as larval stages in fish (close to 45% overall) than for cestodes (≈30%) and nematodes (≈15%). These estimates are very rough, since many parasite genera are found as larvae in some fish species within a family and as adults in others. Still, they suggest that a significant decay relationship would be less likely in trematodes than in the other groups. With respect to adult parasites, the sort of rampant host switching that could neutralize the effect of host phylogenetic distance on patterns of similarity has indeed been detected in cestodes or nematodes parasitizing freshwater fishes (e.g. Skerikova et al. Reference Skerikova, Hypsa and Scholz2001). However, it remains unclear why trematodes should differ from cestodes and nematodes, given that all of these helminths maturing in fish are acquired via ingestion.
A few sources of errors may have generated background noise in the data, possibly contributing to the relatively weak patterns observed. Firstly, the lists of parasite records may have been less complete for some fish species than others. Working at the parasite genus level should have limited any inaccuracy associated with identification errors, taxonomic synonymies or records specifying only the genus of a parasite and not its full species name. However, lists of parasite taxa for each host species may, in part, reflect uneven sampling effort. Inadequate sampling often means that some parasite taxa are missed in field surveys (Walther et al. Reference Walther, Cotgreave, Price, Gregory and Clayton1995; Poulin, Reference Poulin1998, Reference Poulin2004). Excluding fish species with fewer than 3 known parasite genera from the present analyses should have eliminated most of the poorly sampled host species, but some probably remain. Secondly, properties of the host-parasite species-by-genera matrices varied among analyses, and some of these properties, particularly matrix fill, are known to influence the outcome of analyses of matrix properties in biogeographical studies (e.g., Wright et al. Reference Wright, Patterson, Mikkelson, Cutler and Atmar1998). Matrix fill is dependent on host specificity of the parasites and/or richness of the parasite faunas in the fish hosts, and it clearly can affect the slope of the distance decay relationship. For example, in the extreme case where all parasite genera infect all host species, matrix fill is 100%, similarity is maximal between all pairs of host species (all parasite faunas are identical), and the slope of the relationship is zero. Thus, whatever host and parasite properties lead to high matrix fill should homogenize parasite faunas across host species and weaken any decay of similarity as a function of phylogenetic distance. Here, matrix fill did not differ between significant and non-significant regressions of parasite similarity against host phylogenetic distance, and therefore matrix properties probably did not affect the results, Thirdly, the parasite data used here are presence/absence data, and greater discriminatory power would in principle be possible if data on prevalence or intensity of infection were used instead. These measures of infection level, however, vary greatly among localities, even for a particular parasite species in a given host species (Poulin, Reference Poulin2006), and it is not possible to obtain a representative value for any host-parasite taxonomic combination. Fourthly, measures of phylogenetic distances are based on different genes for some fish families. Rates of molecular evolution are not the same in all parts of the genome, and the distance values used here are not fully equivalent among fish families. However, in some of the studies used here, phylogenies of the same fish species based on different genes showed similar topologies as well as branch lengths (Near et al. Reference Near, Bolnick and Wainwright2004; Sloss et al. Reference Sloss, Billington and Burr2004), and thus there is no strong reason to believe that grossly inaccurate phylogenetic distances may have biased the results.
The results of this study provide a bridge between, on the one hand, phylogenetic processes leading to the diversification of parasite faunas and, on the other hand, biogeographical processes that shape local communities. In both cases, the same quantitative pattern of exponential decay of similarity with increasing distance seems to apply. Although not quite universal in biogeographical space, distance decay relationships are known from a wide range of taxa and a range of spatial scales (see Soininen et al. Reference Soininen, McDonald and Hillebrand2007; Morlon et al. Reference Morlon, Chuyong, Condit, Hubbell, Kenfack, Thomas, Valencia and Green2008; Thieltges et al. Reference Thieltges, Ferguson, Jones, Krakau, de Montaudouin, Noble, Reise and Poulin2009 and references therein). In addition, distance decay in the similarity of parasite communities has also been observed as a function of host phylogeographical distance, i.e. by substituting geographical distances between populations of the same host species with genetic distances between them (Seifertova et al. Reference Seifertova, Vyskocilova, Morand and Simkova2008). The present study went one step further, searching for similar patterns at a higher scale, i.e. that between host species. Further studies on parasite assemblages in different host taxa are necessary before we can assess how widespread distance decay relationships are in phylogenetic space. Nevertheless, the results presented here suggest that decreases in community similarity as a function of increasing distance occur in the same manner along any dimension, even if the time scales involved to assemble these communities are vastly different.
I thank B. Krasnov for assistance with the boundary tests, and B. Krasnov, D. Thieltges and J. Timi for comments on an earlier version of the manuscript.